
Multifaceted roles of aerobic glycolysis and oxidative phosphorylation in hepatocellular carcinoma
- Published
- Accepted
- Received
- Academic Editor
- Katherine Mitsouras
- Subject Areas
- Biochemistry, Molecular Biology, Gastroenterology and Hepatology, Oncology
- Keywords
- Hepatocellular carcinoma, Mitochondrial metabolism, Oxidative phosphorylation, Aerobic glycolysis
- Copyright
- © 2023 Zhang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2023. Multifaceted roles of aerobic glycolysis and oxidative phosphorylation in hepatocellular carcinoma. PeerJ 11:e14797 https://doi.org/10.7717/peerj.14797
Abstract
Liver cancer is a common malignancy with high morbidity and mortality rates. Changes in liver metabolism are key factors in the development of primary hepatic carcinoma, and mitochondrial dysfunction is closely related to the occurrence and development of tumours. Accordingly, the study of the metabolic mechanism of mitochondria in primary hepatic carcinomas has gained increasing attention. A growing body of research suggests that defects in mitochondrial respiration are not generally responsible for aerobic glycolysis, nor are they typically selected during tumour evolution. Conversely, the dysfunction of mitochondrial oxidative phosphorylation (OXPHOS) may promote the proliferation, metastasis, and invasion of primary hepatic carcinoma. This review presents the current paradigm of the roles of aerobic glycolysis and OXPHOS in the occurrence and development of hepatocellular carcinoma (HCC). Mitochondrial OXPHOS and cytoplasmic glycolysis cooperate to maintain the energy balance in HCC cells. Our study provides evidence for the targeting of mitochondrial metabolism as a potential therapy for HCC.
Introduction
Cancer is a leading cause of death worldwide and an important barrier to increased life expectancy (Bray et al., 2021). Liver cancer is one of the most common malignant tumours, causing an estimated 906,000 new cases and 830,000 deaths annually worldwide. Primary hepatic carcinoma was the sixth most common cancer, third leading cause of cancer-related deaths worldwide, and leading cause of cancer-related deaths in China among men in 2020. Among primary hepatic carcinoma, hepatocellular carcinoma (HCC) accounts for 75–85% of all cases (Sung et al., 2021). The highest rates of HCC have been documented in East Asia and Africa, with more than one million annual cases projected by 2025 (Llovet et al., 2021). As a highly invasive tumour, HCC is associated with poor prognosis. Although immune checkpoint inhibitors have some therapeutic effect, patient survival time remains short and treatment options are limited.
Mitochondria participate in a variety of metabolic processes, including OXPHOS, calcium and iron homeostasis, apoptosis, and reactive oxygen species production (Andreyev & Starkov, 2005; Boese & Kang, 2021). Their dysfunction is closely related to the occurrence and development of human cancers (Kim, Maiti & Barrientos, 2017). Senescent cells exhibit tumour-friendly behaviour, as evidenced by an increase in mitochondrial calcium load, oxygen consumption rate (OCR), and OXPHOS. These traits help cells escape oncogene-induced senescence (Farfariello et al., 2022). Decades ago, Otto Warburg observed that cancer favoured glucose fermentation even in the presence of oxygen, suggesting that defects in mitochondrial respiration may be the underlying cause of cancer (Warburg, 1956a; Warburg, 1956b). To meet their demand for rapid growth, tumour biomacromolecules can be synthesised using intermediate metabolites generated during aerobic glycolysis (Vaupel, Schmidberger & Mayer, 2019). However, not all tumours share this property of aerobic glycolysis. Furthermore, it is now apparent that defects in mitochondrial respiration are generally not the cause of aerobic glycolysis, nor are they specifically selected during tumour evolution (Zong, Rabinowitz & White, 2016). Several studies suggest that the role of mitochondria in cancer is more complex than that envisioned by Warburg. The mitochondrial 1C enzymes serine hydroxymethyltransferase 2 (SHMT2) and methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) are among the most transcriptionally upregulated genes in cancer (Nilsson et al., 2014; Vazquez, Tedeschi & Bertino, 2013). Tumorigenesis can be impaired by inhibiting the replication of mitochondrial DNA (Tan et al., 2015). Inactivation of the mitochondrial transcription factor Tfam, which depletes mitochondria from tumour cells, disrupts the growth of K-ras lung tumours (Weinberg et al., 2010). These studies emphasise the importance of mitochondrial function in tumour growth.
The liver is not only the largest detoxifying organ but is also critically involved in controlling energy metabolism (Piccinin, Villani & Moschetta, 2019), with the mitochondria as the primary sites of metabolism. Non-alcoholic fatty liver disease, alcoholic fatty liver disease, viral hepatitis, and HCC are closely associated with mitochondrial dysfunction (Zhang et al., 2022; Middleton & Vergis, 2021; Zhang et al., 2019). Reprogramming of mitochondrial metabolism is inextricably linked to the growth and development of HCC (Wang et al., 2021; Wang et al., 2016; Lee et al., 2021). In addition to aerobic glycolysis, OXPHOS plays a key role in HCC cell survival and progression. Unlike for glycolysis, the therapeutic targeting of mitochondrial OXPHOS has received little attention. Based on the most recent findings, this review analyses how aerobic glycolysis and OXPHOS affect the formation, progression, and drug resistance of HCC. Our study makes a significant contribution to the literature, given the global importance of liver cancers and the general lack of comprehensive and tenable theories regarding cancer metabolism. Further, we believe that this article will be of interest to the readership of oncologistsas it highlights the potential avenues for treatment of HCC based on crucial molecules involved in metabolic processes.
Survey Methodology
Relevant published articles were identified from Pubmed, Scopus, Web of Science, China National Knowledge Infrastructure using the search term of “hepatocellular carcinoma”, “mitochondrial metabolism”, “oxidative phosphorylation”, and“aerobic glycolysis.” We performed by crossing these descriptors using the boolean operators “OR” and “AND”. We mainly included relevant articles from 2015 to 2022. Articles related to pyrimidine, amino acid, and phospholipid metabolism with mitochondria and articles not related to hepatocellular carcinoma were excluded. The resulting articles were included where it considered aerobic glycolysis and oxidative phosphorylation in hepatocellular carcinoma. We made a comprehensive interpretation and analysis. To discuss the complex relationship between the two, we added the search term of “L-lactate”. In the process of summarizing the literature on the biological behavior of hepatocellular carcinoma, we added key search terms such as “autophagy” and “angiogenesis” Regarding drug resistance of HCC, we added the search term “sorafenib”. Briefly, the review identified 151 relevant research articles from research labs working on aerobic glycolysis and oxidative phosphorylation in hepatocellular carcinoma.
Aerobic glycolysis during HCC progression and metastasis
In the 1920s, Warburg, Posener & Negelein (1924) found that glycolysis occurred even in the presence of sufficient oxygen in rat liver cancer. Considerable research on aerobic glycolysis in HCC has provided further insights. The ATP produced by aerobic glycolysis may enable HCC to adapt to an energy-deficient tumour microenvironment (Xu & Herschman, 2019). Macromolecules essential for the proliferation of HCC can also be produced through aerobic glycolysis (Heiden, Cantley & Thompson, 2009; Ganapathy-Kanniappan, 2018). Hence, the onset and progression of HCC are associated with enhanced glycolysis (Feng et al., 2020b).
Enhanced expression of aerobic glycolysis enzymes in HCC
Aerobic glycolysis is primarily regulated by hexokinases (HKs), phosphofructokinases (PFKs), and pyruvate kinases (PKs), and the overexpression of these enzymes is associated with a poor prognosis in HCC (Feng et al., 2020b). In this section, we discuss regulators of key glycolytic enzymes to highlight novel therapeutic targets for the treatment of HCC.
The methodological principle is illustrated by a scheme shown in (Fig. 1).
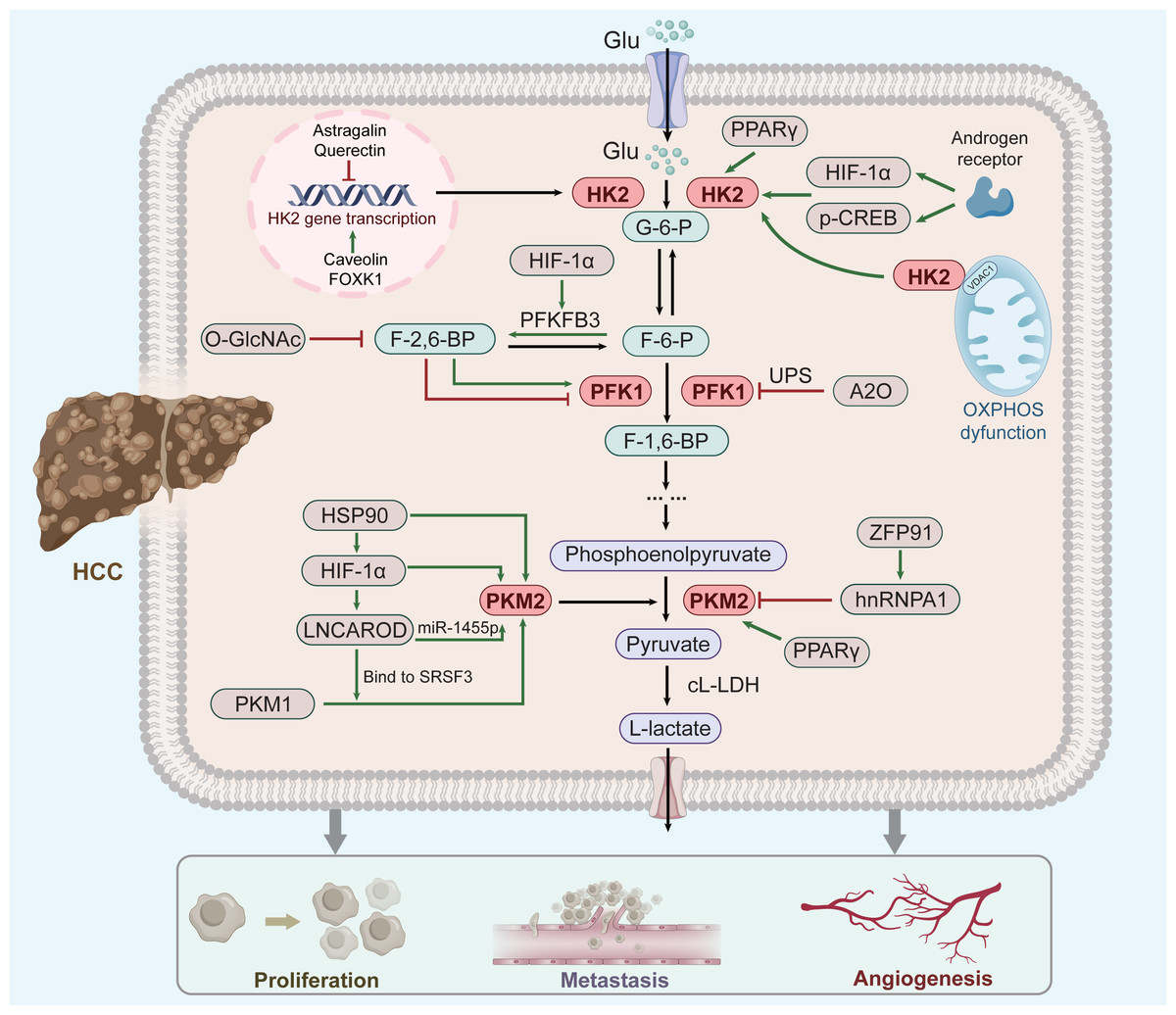
Figure 1: Factor regulation of key enzymes in aerobic glycolysis of HCC.
As three rate-limiting enzymes of glycolysis, HK2, PFK1 and PKM2 play an important role in aerobic glycolysis of HCC cells. These three enzymes are regulated by a number of key factors (green clippings indicate facilitation, red arrows indicate inhibition; see text for details and references). Moreover, their elevated expression can promote the respiratory metabolism of liver cancer to the direction of aerobic glycolysis, thus promoting the occurrence and development of liver cancer.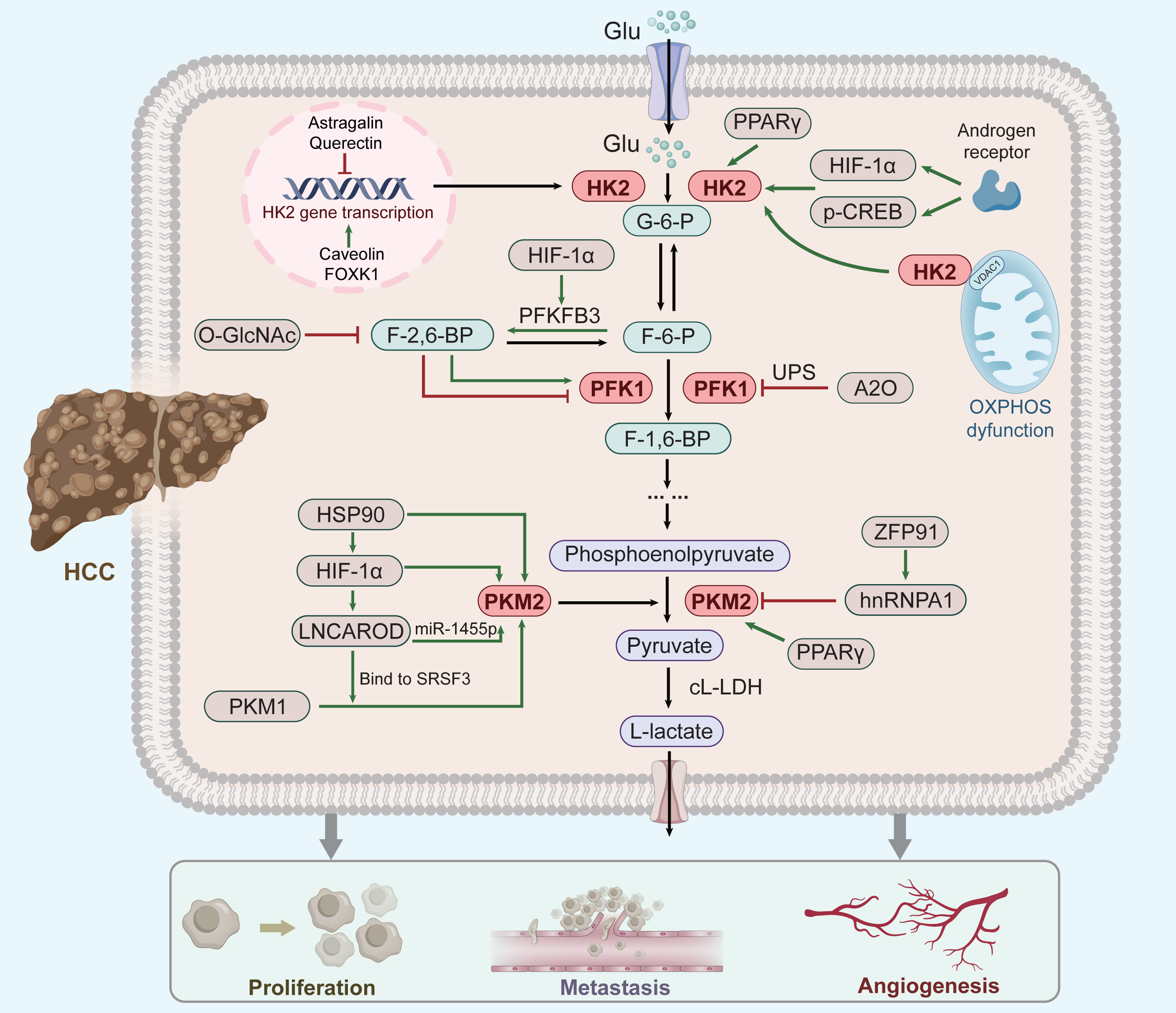{kind=link}
HK2
Overexpression of HK2 leads to a poor prognosis of HCC (Zhang et al., 2016), and increased HK2 levels significantly enhance HCC propagation (Li et al., 2022). Several upstream factors, including Astragalin (Li et al., 2017c), Quercetin (Wu et al., 2019), Caveolin-1 (Chai et al., 2019), and Forkhead box 1(FOXK1) (Cui et al., 2018) regulate HK2 mRNA and protein levels in HCC. Furthermore, FOXK1 and Quercetin act on HK2-dependent glycolysis via the AKT/mTOR pathway (Wu et al., 2019; Cui et al., 2018). Moreover, HK2 binds to voltage-dependent anion-selective channel protein 1(VDAC1) to protect it from the inhibitory effects of downstream products, thereby enhancing glycolysis (Mathupala, Ko & Pedersen, 2009).
The androgen receptor potentiates protein kinase A/cyclic adenosine monophosphate response element-binding (CREB) protein signalling to promote HK2 expression, ultimately promoting HCC cell glycolysis and growth (Sun et al., 2021). This might explain why liver cancer ranks second in male mortality (Sung et al., 2021). Moreover, peroxisome proliferator-activated receptor gamma(PPAR γ) and hypoxia-inducible factor-1 α (HIF-1 α) might contribute to HK2 induction in fatty liver disease and its evolution towards cirrhosis and carcinogenesis (Panasyuk et al., 2012; Mesarwi et al., 2016). Thus, HK2 up-regulation correlates with hepatic diseases progression regardless of cause. (Xu & Herschman, 2019).
PFK1
PFK-1 catalyzes the conversion of fructose 6-phosphate(F-6-P) to fructose 1, 6-bisphosphate(F-1,6-P) and is inhibited by high ATP and phosphoenolpyruvate levels and activated by fructose-2,6-bisphosphate(F-2,6-P) (Okar et al., 2001). PFK-1 is also regulated by posttranslational modifications, such as glycosylation and acetylation (Yi et al., 2012; Feng et al., 2020c). Both hypoxia and glucose deficiency can induce PFK1 glycosylation. O-linked β-N-acetylglucosamine (O-GlcNAc) of PFK1 directly regulates glycolysis to increase cancer cell growth (Yi et al., 2012). Tumor cells express PFK-1 isoforms at high levels to attain a persistent change in glycolytic flux (Ros & Schulze, 2013). In addition, the E3 ubiquitin ligase A20 (A20) promotes the ubiquitination of liver-type phosphofructokinase 1 (PFKL) to downregulate aerobic glycolysis (Feng et al., 2020c). PFKL can mediate multienzyme assembly for glucose metabolism (Kohnhorst et al., 2017), with its tetrameric form affecting glycolysis more stably (Bartrons et al., 2018). The downstream products of PFK1 induce the dissociation of PFK1 tetramers to dimers, leading to the inhibition of PFK1 activity (Wu et al., 2020). In addition, the enzymes 6-phosphofructo-2kinase/fructose-2,6-bisphosphatase-3 (PFK2/PFKFB3) is involved in liver fibrogenesis (Mejias et al., 2020), activates PFK1, and stimulates high glycolytic flux in human cancers (Bando et al., 2005). Metformin inhibits the expression and activity of PFK1 by suppressing PFKFB3 (Zeng et al., 2019). Overall, PFK-1 plays a critical role in the glycolytic pathway.
PKM2
PKM2 is ubiquitously expressed in tumour tissues and regulates cancer cell metabolism (Yang & Lu, 2013; Mazurek, 2011). The expression of PKM2 is controlled by several signaling pathways and transcription factor. HIF-1 α regulates HCC cell glycolysis, and its high expression corroborates the Warburg effect (Feng et al., 2020b). HIF-1 α is the primary upstream regulator of PKM2 in HCC (Feng et al., 2020a), and induces the expression of L-lactate dehydrogenase(L-LDH) and pyruvate dehydrogenase kinase, which promote glycolytic conversion (Kelly & O’Neill, 2015). Heat shock protein 90 (HSP90) promotes the overexpression of HIF-1 α (Liu et al., 2016). In addition, HSP90 also directly regulates the expression of PKM2. Xu et al. (2017) suggested that HSP90 could induce the Thr-328 phosphorylation of PKM2 and increase the abundance of PKM2 in HCC. Thus, the HSP90/HIF-1 α/PKM2 axis has an important effect on aerobic glycolysis. The following reflects another way in which HIF-1 α affects PKM2. HIF-1 α knockdown attenuates the effect of LNCAROD (a newly identified Long non-coding RNA) (Ge et al., 2021). LNCAROD increases PKM2 levels by either combining with serine- and arginine-rich splicing factor 3 (SRSF3) to trigger PKM switching from PKM1 to PKM2, or by sponging miR-1455p to upregulate PKM2 (Jia et al., 2021). However, HCC chemoresistance is also strengthened by LNCAROD overexpression (Jia et al., 2021), although no clear mechanism has been suggested. Additionally, the c-myc-hnRNP A1 pathway has been shown to regulate PKM splicing, increasing expression of PKM2 (David et al., 2010). In HCC, E3 ligase zinc finger protein 91 (ZFP91)—a tumour suppressor gene—regulates alternative splicing of the glycolytic enzyme PKM pre-mRNA through hnRNP A1 to reduce the expression of PKM2 (Chen et al., 2020). PPAR γ also contributes to the pathogenesis or development of hepatic diseases through PKM2 (Panasyuk et al., 2012). Thus, PKM2 activity is regulated by numerous allosteric effectors. Accordingly, targeting PKM2 can be a new therapeutic approach for HCC.
Aerobic glycolytic Micro RNAs involved in HCC
Micro RNAs (MiRNAs) play an important role in aerobic glycolysis of HCC (Nie et al., 2015; Shao et al., 2019; Li et al., 2018a). Several miRNAs, including miR-338, miR-199a-5p, miR-517a, miR-885-5p, miR-129-5p, and miR-3662, are potentially involved in glycolysis and associated with HCC progression. miR-338 can negatively regulate PFKL directly to suppress the Warburg effect. While miR-199a-5p and miR-885-5p inhibit glucose consumption, cell proliferation, and tumorigenesis in HCC cells by targeting the key glycolytic enzyme HK2, miR-517a acts as an oncogene and promotes glycolysis in HCC. MiR-3662 can suppress the Warburg effect and HCC progression by decreasing the expression of HIF-1 α (Zhang et al., 2018; Xu et al., 2019; Chen et al., 2018; Guo et al., 2015; Zheng et al., 2019).
Aerobic glycolytic products are associated with HCC
L-lactate produced by glycolysis is closely related to HCC production (Lee et al., 2017). Overexpression of the peroxisome proliferator–activated receptor γ coactivator 1 α (PPARGC1A, hereafter abbreviated as PGC-1 α) in HCC cell lines suppresses glycolysis and reduces extracellular L-lactate levels (Zuo et al., 2021). Kalhan et al. (2011) reported increased L-lactate production in non alcoholic steatohepatitis (NASH) patients, indicating that glycolytic transformation occurs at the beginning of HCC. Besides bridging glycolysis and OXPHOS, L-lactate has additional functions in cancer cell survival (deBari & Atlante, 2018). L-lactate induces the apoptosis of T and NK cells (San-Millán & Brooks, 2017). Elevated L-lactate levels are associated with early disease metastasis and a short overall and disease-free survival (Cai et al., 2021).
NADH produced by the tricarboxylic acid (TCA) cycle acts as a mitochondrial electron source to initiate electron transfer and produce ATP. Electron flow in glycolysis does not happen through this pathway due to a lack of oxygen as an electron acceptor. This abnormal electron flow in the electron transport chain generates a large amount of reactive oxygen species (ROS)(Chiu et al., 2019). An increased ROS response can activate the mitogen activated protein kinase (MAPK) signalling pathway (Win et al., 2015; Win et al., 2016). MAPK activates PGC-1 α, which is a key regulator of mitochondrial metabolism (Rabinovitch et al., 2017; Wang et al., 2012) and an important regulatory target of sirtuin-1 (SIRT1) (Li et al., 2016), which can dynamically affect different types of tumours (Firestein et al., 2008; Sun et al., 2013). SIRT1 is up-regulated in HCC and facilitates tumour invasion and migration by activating the SIRT1/PGC-1 α axis.
Two primary effects of OXPHOS in HCC
OXPHOS role in the transformation of hepatitis into HCC
Under normal circumstances, the OXPHOS system is critical for energy production and cell survival. Many genes involved in OXPHOS are downregulated in chronic hepatitis, especially in DNA methyltransferase 3B (DNMT3B) deficient chronic hepatitis, which is a key component in DNA methylation (Lyko, 2018). Low levels of OXPHOS in chronic hepatitis promotes fibrosis, sclerosis, and carcinogenesis (Iguchi et al., 2020). Additionally, activation of hepatitis C virus replication down-regulates respiratory chain complex I and IV activity in an HCC cell line. The ensuing metabolic alterations in the pentose phosphate and glycolysis pathways reprogramme the cells to develop HCC (Gerresheim et al., 2019). Hence, defects in OXPHOS can shift the inflammatory status of the liver to promote tumour formation (Iguchi et al., 2020; Gerresheim et al., 2019).
The OXPHOS defect persists after the formation of HCC cells, thereby allowing HCC cells to survive and become more malignant. For example, high expression of DNMT3B in HCC predicts poor prognosis (Oh et al., 2007). C-Src is one of the most important Src family kinases (SFKs), and its overexpression inhibits the expression of the nuclear and mitochondrial coding subunits of OXPHOS complexes I and IV in HCC (Hunter, Koc & Koc, 2020). Zhao et al. (2015) showed that total Src was closely associated with a poor clinical prognosis in HCC. The SFK inhibitor PP2 can restore the c-Src-mediated OXPHOS damage and inhibit HCC cell growth to a large degree (Hunter, Koc & Koc, 2020). Furthermore, an increased expression of claudin-1, an oncogenic factor associated with poor prognosis (Chen et al., 2017; Suh et al., 2013) , has been observed in highly invasive OXPHOS-deficient HCC cells (Kim et al., 2011). The down-regulation of mitochondrial ribosomal protein L13 (MRPL13) results in OXPHOS deficiency and promotes the invasiveness of HCC cells by inducing claudin-1 expression (Lee et al., 2017).
Moreover, the mitochondrial fission and fusion imbalance also contributes to cancer (Chan, 2020). Mitochondrial fusion can increase OXPHOS enzyme levels and the recovery of OXPHOS levels reduces proliferation and metastasis in HCC (Zhang et al., 2020). The dynamin superfamily of GTPases can promote mitochondrial fusion (Chan, 2020). RAS-associated binding 3A protein (Rab3A), a small Ras-like GTPase, stimulates mitochondrial OXPHOS activity and suppresses HCC metastasis (Wu et al., 2018).
In summary, OXPHOS deficiency can promote the development of HCC. This changes the long-held view that OXPHOS disruption is an epiphenomenon of tumours. The current evidence provides an impetus for further research to uncover related mechanisms.
OXPHOS enhances the malignant behavior of HCC
Tumour cells without mitochondrial DNA cannot undergo OXPHOS or maturation, which suggests that OXPHOS is critical for cancer cell survival (Tan et al., 2015). Energy synthesis and nucleotide biosynthesis during cancer cell proliferation require sufficient ATP to provide energy (Sullivan et al., 2015; Birsoy et al., 2015). The survival of tumour cells with mutant β-catenin relies on the production of ATP by mitochondrial OXPHOS (Shikata et al., 2017). Moreover, blood supply to HCC cells mainly originates from the hepatic artery, which provides sufficient oxygen for OXPHOS (Sezai et al., 1993). These findings imply a close relationship between elevated OXPHOS levels and HCC progression. Interestingly, ATP production in HepG2 cells is considerably limited when only mitochondrial OXPHOS is inhibited, confirming the importance of OXPHOS in HepG2 cells (Yang et al., 2020). Liver Cancer Stem Cells from HCCLM3 cells relies on OXPHOS to enhance its malignant biological behaviors (Liu et al., 2020). Hoki et al. (2019) showed that HCC patients with low divalent metal-ion transporter-1 (DMT1) expression—corresponding to high OXPHOS levels—had shorter disease-free survival rates. Further in vitro and in vivo studies are needed to validate these findings and reveal how DMT1 affects the mitochondrial respiratory processes. Li et al. (2017a) showed that mitochondrial elongation facilitates the formation of cristae and assembly of the respiratory system in HCC cells under energy stress. This indicates a shift in the metabolic pathway from glycolysis to OXPHOS, benefitting HCC cell survival in vitro and in vivo, and a poor prognosis in HCC patients (Li et al., 2017a).
Interpretation of metabolic flexibility in HCC
We further discussed the contradiction between the above two aspects. Normal tissues utilize 90% of ATP produced by OXPHOS and 10% by glycolysis. On the contrary, tumors produce 50% of the ATP from OXPHOS and 50% from glycolysis (Warburg, 1956a). This is despite the fact that OXPHOS is reduced in tumour cells compared to normal cells. However, many tumours maintain a large amount of OXPHOS to produce large amounts of ATP. Hence, a finely tuned intrinsic mechanism exists to maintain OXPHOS and mitochondrial function, helping HCC cells to select metabolic mode. HepG2 cells had increased mitochondrial content, OXPHOS levels, and decreased glycolysis levels under aglycemic conditions than under hyperglycemic condition (Domenis et al., 2012). This showed that the energy substrate type has a certain influence on the metabolic mode selected by HCC cells. In addition, ubiquinol-cytochrome c reductase complex assembly factor 3 (UQCC3) is a mitochondrial protein and a human complex III assembly factor (Desmurs et al., 2015). In hypoxia, it can simultaneously sustain OXPHOS, and glycolytic activity (Yang et al., 2020). The enhancement of OXPHOS maintained mitochondrial function at a necessary level to support HCC cells adapting to hypoxic stress. It reflected that HCC can exhibit metabolic flexibility. Current evidence indicates that mitochondrial metabolism in HCC is not impaired. However, to date, the molecular mechanisms underlying the choice between glycolysis and OXPHOS during energy stress in HCC cells are not well understood. Existing experimental techniques may not be able to adequately reflect the level of OXPHOS, so it is important to use a variety of experimental methods to fully verify the activity of OXPHOS. Further studies are required to understand the relationship between OXPHOS and HCC development.
The complex relationship between aerobic glycolysis and OXPHOS HCC
OXPHOS levels are strongly correlated with the levels of L-lactate and pyruvate. The p53 up-regulated modulator of apoptosis (PUMA)-mediated apoptotic response in hepatocytes is a direct cause of compensatory proliferation and ensuing carcinogenesis (Qiu et al., 2011). Wild-type p53 (WTp53) promotes glycolysis by activating PUMA and inhibiting pyruvate-driven OXPHOS in HCC patients (Kim et al., 2019). Moreover, overexpression of PTEN restores the inactivated PI3K/Akt pathway, significantly inhibits L-lactate production, and enhances access of pyruvate to mitochondria (Zhao et al., 2020). Hence, efficient glycolysis promotes L-lactate accumulation, thereby ensuring a reduced mitochondrial pyruvate intake and preventing normal OXPHOS processes.
However, glycolysis seems to play a different role in HCC cells that depend on OXPHOS for survival: L-lactate, but not pyruvate, is thought to be the end product of glycolysis and the main substrate of the mitochondrial TCA cycle (see Fig. 2) (deBari & Atlante, 2018; Schurr, 2018). L-lactate appears to be the favoured substrate during high metabolic activity in the heart and brain, where it is involved in mitochondrial oxidation processes (Schurr, 2018; Passarella et al., 2008). ATP production in HepG2 cells greatly depend on OXPHOS (Yang et al., 2020). OXPHOS may be promoted by an increase in the extracellular L-lactate output from neighbouring glycolytic tumour cells (Hong et al., 2019; Schurr & Passarella, 2022) Mitochondrial L-lactate dehydrogenase (mL-LDH) is highly expressed in Hep G2 cell, where pyruvate carrier function is diminished. This suggests that pyruvate metabolism may be not the main driver of OXPHOS activity in Hep G2 cells (Pizzuto et al., 2012). mL-LDH can oxidise L-lactate, leading to low OCR and membrane potential generation. Pyruvate is also formed in this process and continues to participate in the TCA cycle (deBari & Atlante, 2018; Passarella et al., 2008; Pizzuto et al., 2012; Passarella & Schurr, 2018). This L-lactate mechanism can restore the L-lactate/pyruvate shuttle and the key substances, oxaloacetate (OAA), malate (MAL), and citrate (CIT) in Hep G2 cells, also appear outside the mitochondria because of L-lactate uptake and metabolism (Pizzuto et al., 2012; Passarella & Schurr, 2018). The L-lactate/pyruvate shuttle can compensate for the limited ability of cancer cells to recycle NADH to NAD+ (deBari & Atlante, 2018).
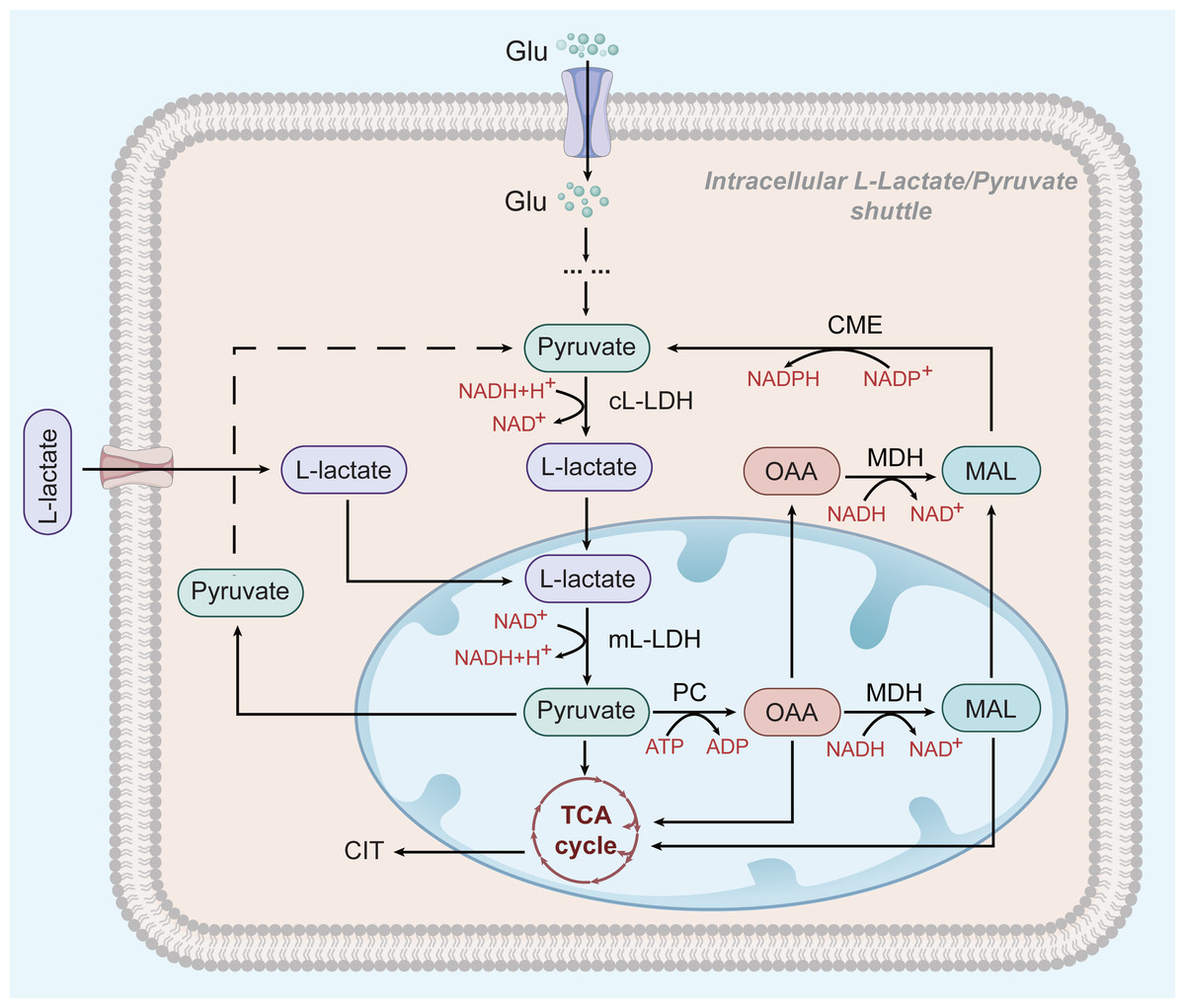
Figure 2: L-lactate enters mitochondria of HCC and participates in OXPHOS.
In HCC cells, L-lactate produced by aerobic glycolysis can enter mitochondria and participate in the OXPHOS process as a substrate. After entering the mitochondria, it is oxidized by mL-LDH to pyruvate, which subsequently produces OAA and MAC. Pyruvate, OAA, and MAL can be exported to the cytoplasm to be used for the L-lactate/pyruvate shuttle.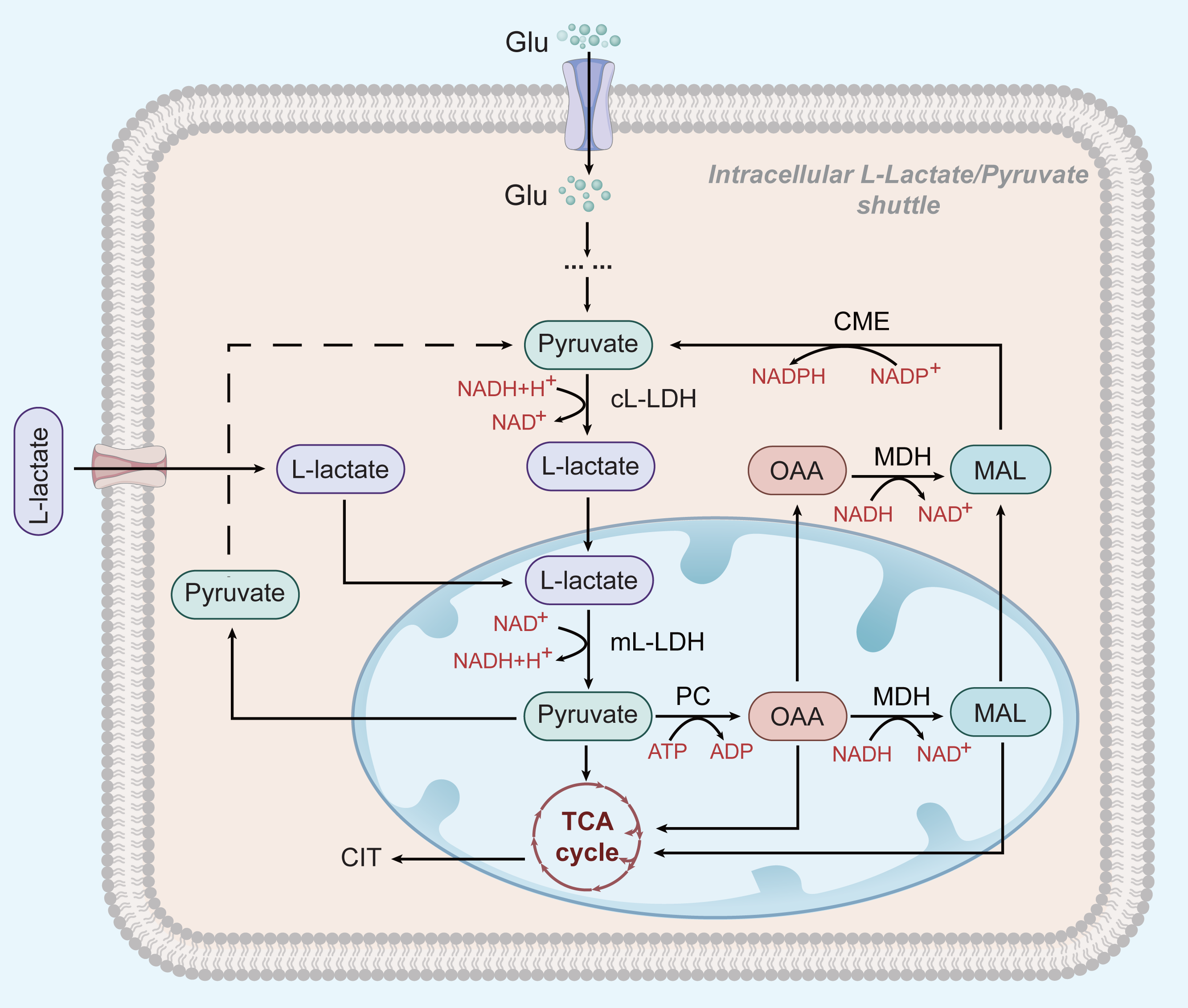{kind=link}
The above shows that L-lactate participates in the transport of essential molecules from the cytosol to the mitochondria and promotes the OXPHOS process in Hep G2 cells. However, Lee et al. (2017) noted that defects in OXPHOS may be triggered by an increased extracellular L-lactate release from the surrounding glycolytic tumour cells. Taken together, the results of these studies are contradictory. Future studies should explore the mechanism of L-lactate/pyruvate transport into mitochondria as a target to disrupt the relationship between glycolysis and OXPHOS and suppress tumour growth.
The available information seems to indicate that key steps in both glycolysis and OXPHOS pathways must be simultaneously inhibited to effectively kill HCC cells through energy deprivation. However, a logical explanation for this thermodynamic resistance is lacking. The significance and consequences of fluctuations in lactate levels in HCC are poorly documented, requiring further research on the intricacies of HCC metabolism.
Links between mitochondrial metabolic reprogramming and viable biological processes in hepatocellular carcinoma
Autophagy
The occurrence and progression of tumours are closely linked to autophagy—a process in which dysregulated and damaged cells (e.g., mitochondria) get surrounded by vesicles that produce autophagosomes, which are eventually degraded by lysosomes (Ashrafi & Schwarz, 2013; Jiang & Mizushima, 2014). Autophagy has the following two roles in HCC: when oncogene activation and cell cycle regulation are lost in the early stages of liver damage, autophagy seems to be a tumour suppressor; however, after altering the metabolism of cancer cells and blocking the apoptotic pathway, autophagy appears to promote cancer development (Bartolini et al., 2018; Harper, Ordureau & Heo, 2018; Panigrahi et al., 2020). Autophagosome formation is induced in hypoxic tumor regions (Song et al., 2009). Autophagy also plays a key role in glycolysis by increasing glucose consumption and L-lactate production, thereby promoting glycolysis in HCC (Fan et al., 2018). Overexpression of the transcription repressor chicken ovalbumin upstream promoter-transcription factor 2 (COUP-TFII) leads to mitochondrial dysfunction (Kao et al., 2020). Under hypoxic conditions, histone deacetylase 6 (HDAC6) activity increases and anti-acetyl- β-catenin (Lys49) deacetylation mediated by HDAC6 enhances the nuclear translocation of β-catenin and its binding to COUP-TFII. Additionally, HDAC6 reduces OXPHOS through the β-catenin/COUP-TFII pathway induced by autophagy in HCC (Yan et al., 2021). Notably, autophagy induces monocarboxylate transporter 1 (MCT1) expression by activating the Wnt/ β-catenin signalling pathway, thereby promoting glycolysis and HCC metastasis (Fan et al., 2018). These findings demonstrate a close connection between autophagy and the metabolic status of HCC. However, autophagy supports OXPHOS in acute myeloid leukaemia by supplying free fatty acids through lipid catabolism (Bosc et al., 2020). Therefore, it is possible that distinct cancer types have different autophagy-regulating mechanisms to control metabolism. Further studies are needed to investigate the mechanism of autophagy in HCC.
Angiogenesis
Tumour growth can be clinically restricted by blocking tumour blood supply, for example via by transcatheter arterial embolisation or chemoembolisation (Semenza, 2012). The occurrence of glycolysis is closely related to angiogenesis, possibly due to the role of L-lactate in carcinogenesis, specifically angiogenesis (Brooks, 2018). Vascular endothelial growth factor (VEGF) is released by hypoxic tissues and is highly secreted in the acidified environment (Shi et al., 2001; Eichmann & Simons Coicb, 2012). Overexpression of PKM2 promotes the production of VEGF (Zhang et al., 2021). Additionally, VEGF enhances glycolysis by increasing glucose uptake and driving the expression of glycolysis activators, including PFKFB3 (DeBock et al., 2013). Overall, tissue environment and function are key determinants of VEGF metabolic activity. Fibroblast growth factor receptor (FGFR) is another crucial factor that induces angiogenesis (Liu et al., 2021a). In FGFR-overexpressing tumour cells, L-lactate dehydrogenase A, PFKL, and PKM2 are abnormally upregulated, indicating that glycolysis can promote FGFR amplification (Jin et al., 2019). Fu et al. (2018) showed that PI3K/Akt promoted angiogenesis in HCC. This signalling pathway enhanced glycolytic activity mediated by HK2, PFK1, and PFK2 (Robey & Hay, 2006). Thus, inhibition of glycolytic processes effectively inhibits angiogenesis in HCC. In contrast, in HCC cells lacking HIF-1 α, PI3K-Akt signalling can promote tumour growth by increasing VEGF expression and angiogenesis, which does not involve glycolysis (Arsham et al., 2004). Therefore, the effect of PI3K/Akt signalling on glycolysis in HIF-1 α-deficient tumours under hypoxia needs to be explored.
Taken together, glycolysis promotes HCC progression by regulating signal transduction and mitochondrial biogenesis in HCC cells.
Roles of OXPHOS in HCC treatment
The regulation of OXPHOS through targeted drug treatments has had limited success in HCC. Recovery of OXPHOS can increase the sensitivity of HCC cells to drugs (Plecita-Hlavata, Jezek & Jezek, 2015; Schmidt et al., 2021; Skolik et al., 2021). Sorafenib is the first-line targeted therapy for advanced HCC (Qin et al., 2021; Cheng et al., 2020). The glycolytic activity in HCC cells is strengthened in the presence of sorafenib and promotes sorafenib resistance (Zhang et al., 2017). Spitz et al. (2009) demonstrated that acetylsalicylic acid can promote anti-tumour effects by inhibiting glycolysis and elevating OXPHOS levels. Further studies showed that acetylsalicylic acid reduced glycolysis rates by PFKFB3 in sorafenib-resistant HCC cells, corresponding to a change from glycolysis to OXPHOS. The combined use of sorafenib and acetylsalicylic acid can reduce the proliferation and increase the apoptosis of HCC cells (Li et al., 2017b). Similarly, simvastatin can inhibit PKM2-mediated glycolysis, re-sensitizing HCC cells to sorafenib (Feng et al., 2020a). Therefore, combinatorial therapies with sorafenib are promising and require further investigation.
In addition, OXPHOS also has an impact on the immunotherapy of HCC. Inhibition of OXPHOS in activated T cells inhibits their proliferation and upregulate the genes associated with T cell depletion (Vardhana et al., 2020). Up-regulation of OXPHOS promotes metabolic reprogramming, which restores T cell function and enhances the efficacy of cancer immunotherapy (Guo et al., 2021). Human leukocyte antigen class-I (HLA-I) promotes the immune response by presenting antigenic peptides to cytotoxic T cells (Garcia-Lora, Algarra & Garrido, 2003). HK2 can lead to the downregulation of HLA-I to achieve immune suppression in HCC (Liu et al., 2021b).
Contrary to the above-mentioned view, OXPHOS inhibitors, which lower the OCR and inhibit tumour growth, could be effective agents in anticancer therapies. Aglycemia increases fermentative glycolytic substrate-level phosphorylation following glucose refeeding as well as the responsiveness of both fermentation and OXPHOS to meet the energy demand in HepG2 cells. Hence, glycaemic OXPHOS HepG2 cells exhibit increased drug resistivity (Plecita-Hlavata, Jezek & Jezek, 2015). In addition, HepG2 cells increase their resistance to chloroethylnitrosourea (CENU) by boosting OXPHOS and ATP levels (Loiseau et al., 2009). This seems to contradict the idea that recovery of OXPHOS can increase the sensitivity of HCC cells to drugs. 3-Bromopyruvic acid (3-BrPA)—a potential anti-tumour agent—not only inhibits glycolysis in HepG2 cells but also inhibits the metabolism of glucose and glutamine in the TCA cycle (Li et al., 2018b). Its dual effects provide great value for future exploration of OXPHOS targeted therapy for HCC.
These findings highlight potential strategies to promote the sensitivity of HCC to drugs. We conclude that OXPHOS plays an important role in the sensitivity and resistance to HCC treatment. However, further in vivo studies are required to determine their efficacy, pharmacokinetics, dosing methods, and side effects of such strategies (Ashton et al., 2018).
Conclusions
Cancer was previously believed to be a proliferative disease. However, growing evidence suggests that it is a metabolic disease (Ashton et al., 2018; Roth et al., 2020; Lyssiotis & Kimmelman, 2017; Martinez-Reyes & Chandel, 2021). Dysregulation of cellular metabolism is as widespread in cancer cells as other hallmarks of cancer function (Roth et al., 2020). Metabolic reprogramming is a crucial microenvironmental adaptation in HCC cells (Shi et al., 2009). The occurrence of HCC is highly dependent on elevated levels of aerobic glycolysis. However, HCC, especially the lower differential and metastasis potential cells, are more dependent on OXPHOS. Elevated aerobic glycolysis and decreased OXPHOS promote the development of HCC, and elevated OXPHOS levels have a similar effect. The dynamic regulation mechanism of mitochondria is very complex. There are different regulatory mechanisms under different environmental pressures. Glycolysis and OXPHOS are complex metabolic processes that affect the development, growth, metastasis, drug resistance of liver cancer through a variety of signalling pathways (Table 1). Understanding the molecular mechanisms of mitochondrial activity/silencing in HCC cells and the limitations of this phenomenon is critical for HCC treatment and drug efficacy. Therefore, we still need to explore how OXPHOS and glycolysis can coordinate the metabolic adaptations of HCC cells in the future. We hope to enhance the therapeutic efficacy of HCC by strategically combining multiple drugs that target different metabolic pathways.
| Agent | Target ormechanism | Outcome of HCC | Reference |
|---|---|---|---|
| Astragalin-OE | Upregulates miR-125b to inhibit HK2 | HCC proliferation in vitro | Li et al. (2017c) |
| Quercetin-OE | Reduces HK2 mRNA and protein expression | Inhibits proliferation | Wu et al. (2019) |
| Caveolin-1-OE | Enhances HK2 gene and protein expression | Promotes cellular metabolism, invasion, and migration | Chai et al. (2019) |
| FOXK1-KD | Inhibits HK2expression at both mRNA and protein levels | Supresses HCC cell viability | Cui et al. (2018) |
| 125I irradiation | UpregulatesmiR-338 to inhibit PFKL | Inhibits proliferation and metastasis | Zheng et al. (2019) |
| A20-OE | Downregulates PFKL at the post-transcriptional levels | Inhibits proliferation, clone formation, and metastasis | Feng et al. (2020c) |
| HSP90-OE | Increases PKM2 levels | HCC proliferation and apoptosis | Xu et al. (2017) |
| ZFP91-KD | Regulates the PKM pre-mRNAthrough hnRNP A1to form higher PKM1 isoforms and supresses lower PKM2 isoform | Suppresses proliferation, and metastasis | Chen et al. (2020) |
| LNCAROD-OE | Binds to SRSF3, induces PKM switch towards PKM2; maintain PKM2 by against miR-145-5P | Promotes growth, migration, invasion and chemoresistance. | Jia et al. (2021) |
| c-Src-OE | Impairs the expression of OXPHOS complexes I and IV | Promote proliferation | Zhao et al. (2015) |
| Mfn1-OE | Promotes mitochondrial fusion to induce OXPHOS, and promotes the expression of OXPHOS enzymes | Inhibits proliferation and metastasis | Zhang et al. (2020) |
| Rab3A-OE | Promotes the expression of several coxs to enhance OXPHOS | Attenuates metastasis | Wu et al. (2018) |
| MRPL13-OE | Leads to OXPHOS defects Via CLN1expression | Enhances invasion | Lee et al. (2017) |
| WTp53-OE | Induces PUMA to boost glycolysis and suppress pyruvate-driven OXPHOS | Poor prognosis | Kim et al. (2019) |
| UQCC3-OE | Sustains mtOXPHOS, HIF1a stabilization, and glycolytic activity | More progressive outcomes and shortened survival | Yang et al. (2020) |
Notes:
- OE
-
overexpression
- KD
-
knockdown
- HK2
-
hexokinase2
- PFK1
-
phosphofructokinase
- PFKL
-
Liver-type phosphofructokinase
- PKM
-
pyruvate kinase M
- PKM1
-
pyruvate kinases M1
- PKM2
-
Pyruvate kinase isoform M2
- FOXK1
-
Forkhead box 1
- A20
-
E3 ubiquitin ligase A20
- HSP90
-
Heat shock protein 90
- ZFP91
-
E3 ligase zinc finger protein 91
- c-SrC
-
a member of Src family kinases
- Mfn1
-
mitochondrial fusion protein mitofusin-1
- Rab3A
-
RAS-associated binding 3A protein
- MRPL13
-
mitochondrial ribosomal protein L13
- WTp53
-
Wild-type p53
- UQCC3
-
Ubiquinol-cytochrome c reductase complex assembly factor 3