
DISCo-microbe: design of an identifiable synthetic community of microbes
- Published
- Accepted
- Received
- Academic Editor
- Xavier Harrison
- Subject Areas
- Bioinformatics, Microbiology
- Keywords
- Constructed community, Microbiome, 16S rRNA, Synthetic community, Taxonomic profiling, In vivo experimentation
- Copyright
- © 2020 Carper et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2020. DISCo-microbe: design of an identifiable synthetic community of microbes. PeerJ 8:e8534 https://doi.org/10.7717/peerj.8534
Abstract
Background
Microbiomes are extremely important for their host organisms, providing many vital functions and extending their hosts’ phenotypes. Natural studies of host-associated microbiomes can be difficult to interpret due to the high complexity of microbial communities, which hinders our ability to track and identify individual members along with the many factors that structure or perturb those communities. For this reason, researchers have turned to synthetic or constructed communities in which the identities of all members are known. However, due to the lack of tracking methods and the difficulty of creating a more diverse and identifiable community that can be distinguished through next-generation sequencing, most such in vivo studies have used only a few strains.
Results
To address this issue, we developed DISCo-microbe, a program for the design of an identifiable synthetic community of microbes for use in in vivo experimentation. The program is composed of two modules; (1) create, which allows the user to generate a highly diverse community list from an input DNA sequence alignment using a custom nucleotide distance algorithm, and (2) subsample, which subsamples the community list to either represent a number of grouping variables, including taxonomic proportions, or to reach a user-specified maximum number of community members. As an example, we demonstrate the generation of a synthetic microbial community that can be distinguished through amplicon sequencing. The synthetic microbial community in this example consisted of 2,122 members from a starting DNA sequence alignment of 10,000 16S rRNA sequences from the Ribosomal Database Project. We generated simulated Illumina sequencing data from the constructed community and demonstrate that DISCo-microbe is capable of designing diverse communities with members distinguishable by amplicon sequencing. Using the simulated data we were able to recover sequences from between 97–100% of community members using two different post-processing workflows. Furthermore, 97–99% of sequences were assigned to a community member with zero sequences being misidentified. We then subsampled the community list using taxonomic proportions to mimic a natural plant host–associated microbiome, ultimately yielding a diverse community of 784 members.
Conclusions
DISCo-microbe can create a highly diverse community list of microbes that can be distinguished through 16S rRNA gene sequencing, and has the ability to subsample (i.e., design) the community for the desired number of members and taxonomic proportions. Although developed for bacteria, the program allows for any alignment input from any taxonomic group, making it broadly applicable. The software and data are freely available from GitHub (https://github.com/dlcarper/DISCo-microbe) and Python Package Index (PYPI).
Background
Multicellular eukaryotes live in association with complex communities of microorganisms (Zilber-Rosenberg & Rosenberg, 2008; Bordenstein & Theis, 2015; Rosenberg & Zilber-Rosenberg, 2016) that play important roles in host health and function (Huttenhower et al., 2012; Schlaeppi & Bulgarelli, 2015; Engel et al., 2016). Given the complexity of these systems and our inability to track and identify all members, it is often difficult to disentangle the factors influencing the structure and interactions among host-associated microbiomes. The development of synthetic model communities is a key strategy for addressing this issue (Busby et al., 2017). Next-generation sequencing of marker genes has demonstrated that both abiotic and biotic factors structure host-associated microbiomes (Spor, Koren & Ley, 2011; Huttenhower et al., 2012; Ofek-Lalzar et al., 2014; Adair & Douglas, 2017); however, the marker genes commonly used in these studies provide low taxonomic resolution, making it difficult to identify all microbes present in the community (Caporaso et al., 2011). Metagenomics studies provide insight into potential microbial function, but are not feasible for microbiomes within host tissues due to the presence of excess host DNA (Jiao et al., 2006; Feehery et al., 2013; Thoendel et al., 2016; Marotz et al., 2018). Accordingly, recent studies have utilized synthetic or simplified microbiome approaches to examine the drivers of host-associated microbiome assembly, interactions, and function (Bodenhausen et al., 2014; Lebeis et al., 2015; Timm et al., 2016; Niu et al., 2017). This approach involves adding previously characterized microbial strains to an axenic host organism, allowing for the investigation of colonization, shifts in community structure (Bodenhausen et al., 2014), microbe–microbe interactions, and host–microbe interactions. When such data are paired with genomic information, it becomes feasible to infer microbial strain metabolic potential. Despite the increased use and prioritization of synthetic systems by the research community (Busby et al., 2017), we currently lack adequate methods for systematically designing a microbial community that is identifiable by common sequencing techniques.
Until now, synthetic communities have been constructed from a functional perspective or with limited strains. For example, some researchers have focused on functional assets (characteristics) of microbes to create a specific metabolic output, often by combining a few bacterial (Shong, Jimenez Diaz & Collins, 2012; Mee et al., 2014; Shi et al., 2017) or fungal strains (Minty et al., 2013; Hu et al., 2017). Although useful for bio-engineering purposes, this approach is not as applicable to studies of microbiomes, in which diversity is much greater. Host-associated synthetic communities have also been restricted to a few strains, with confirmation through re-isolation, limiting researchers’ ability to extrapolate to more diverse communities (Bodenhausen et al., 2014; Niu et al., 2017; Herrera Paredes et al., 2018). Recent studies have linked host-associated microbiome function to microbial diversity (Turnbaugh et al., 2008; Laforest-Lapointe et al., 2017), requiring the incorporation of phylogenetic distance into synthetic community design. The design of phylogenetically diverse communities is associated with at least two major challenges: (1) creating a diverse community that can easily be distinguished through common high-throughput sequencing technologies, and (2) ensuring that community members possess the desired attributes (e.g., taxonomic composition and metabolic potential). Without advanced computational abilities, overcoming these challenges is formidable and time-consuming. Furthermore, manual bioinformatic workflows are difficult to document and error-prone, costing additional time and decreasing reproducibility.
In this paper, we describe an easy-to-use command-line program, Design of an Identifiable Synthetic Community of Microbes (DISCo-microbe), for creation of diverse communities of organisms that can be distinguished through next-generation sequencing technology for use in in vivo experiments. DISCo-microbe consists of two modules, create and subsample. The create module constructs a highly diverse community at a specified sequence difference from an input of aligned DNA/RNA sequences, e.g., 16S sequence. The module can either design a de novo community or design a community that includes targeted organisms. create solves problem (1) by easily generating a diverse community of members through an easily documentable method, ensuring reproducibility. The subsample module provides options for dividing the community into subsets, according to either the number of members or the proportions of a grouping variable, both of which can be specified by the user. subsample module solves problem (2) by allowing the user to subsample an already distinguishable community of members based on attributes of interest. Although this software was designed for construction of microbial communities, any DNA/RNA alignment can be used as input; consequently, users are not restricted to any particular organismal group or marker gene. DISCo-microbe is implemented in Python and is available through GitHub and PYPI.
Materials and Methods
DISCo-microbe is a command-line program written in Python and requires Biopython (Cock et al., 2009), which is automatically installed along with the program. We chose to implement DISCo-microbe in Python for easy portability to almost all systems. DISCo-microbe consists of two modules, create and subsample. We have written extensive documentation for DISCo-microbe following the principles outlined in (Seemann, 2013; Karimzadeh & Hoffman, 2018) including a quickstart tutorial that walks users through all commands, illustrating the ease of use and reproducibility of DISCo-microbe.
Workflow
Create module
The create module has two required arguments, an alignment of DNA or RNA sequences in FASTA format (–i-alignment) and a user-specified minimum sequence distance between community members (–p-editdistance). The module uses a greedy algorithm to construct a community maximizing the number of members at the user-specified sequence distance. The optional arguments for the create module include: (i) a community starter list (–p-include-strains), containing members the user would like to be included in the community; (ii) a seed number (–p-seed), for reproducibility; (iii) a metadata file (–i-metadata) for combination with the final community; iv) an option to output the FASTA file (–o-fasta) of the final community and; (v) an option to import a sequence distance database (–i-distance-database; described below).
The create module operates in two distinct phases. The first phase creates a database of all pairwise sequence distances from the input alignment, calculated using a modified Hamming distance. The Hamming distance is a coding theory metric that measures the number of positions at which two sequences of equal length differ. Because the Hamming distance does not consider the nature of the differences, it can be problematic to determine the distance between molecular sequences, in which nucleotide ambiguities can be common; such ambiguities artificially inflate the number of differences between sequences, possibly causing the final community to be less distinguishable than expected (Fig. 1). To deal with IUPAC nucleotide ambiguities, we created a custom Hamming distance, termed the nucleotide Hamming distance, which accommodates nucleotide ambiguities and adjusts the distance value accordingly (Fig. 1). Furthermore, this metric can mitigate sequence errors introduced by PCR and sequencing technologies (Pfeiffer et al., 2018; Filges et al., 2019), allowing the identification of sequences containing up to d − 1 errors, where d is the user-specified minimum sequence distance. Lastly, we included an export of the distance database as a flat file for easy manipulation with command line utilities. This option also allows the user to load the database of previously calculated distances if a modification to the run parameters is wanted. Furthermore, the distance database is updated in real-time as distances are calculated, acting as a checkpoint to resume calculations with minimal lost time in the event that DISCo-microbe quits unexpectedly.
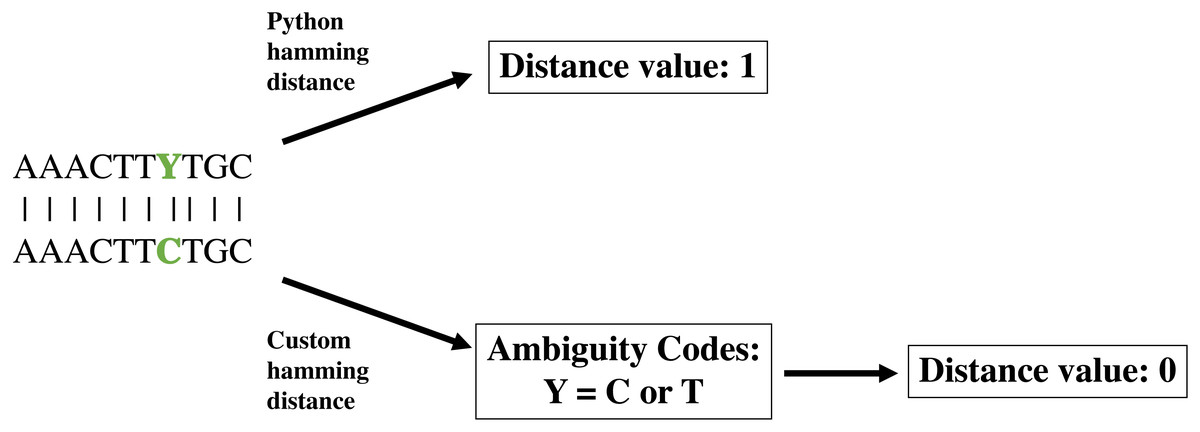
Figure 1: Demonstration of custom nucleotide Hamming distance.
Demonstration of Python Hamming distance and custom nucleotide Hamming distance, which takes into account nucleotide ambiguities.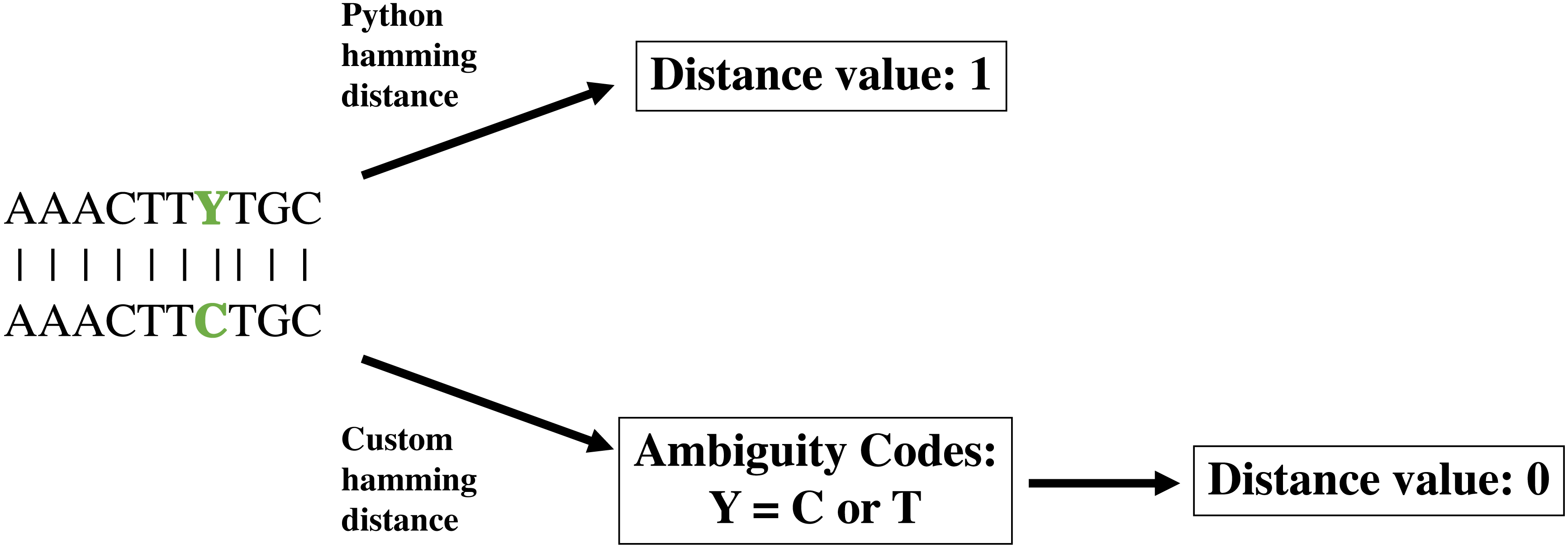{kind=link}
The second phase of the create module runs a greedy algorithm to construct a community. To initiate the community-building algorithm, the user can specify a starting community, which will be validated to determine that all pairwise distances meet the minimum requirement indicated by –p-editdistance. If the starting community is not valid at the indicated sequence distance, an error message with the conflicting sequence identifiers will be displayed. If a starting community is not specified, the individual with the fewest connections at the user-specified sequence distance (–p-editdistance) will be used to initiate the community (Fig. 2). If there is a tie for the fewest connections, one individual is selected at random. Once an initial community is established, the algorithm will iteratively add new members to the community by creating a list of possible members that meet a single requirement. The individual must meet the minimum sequence distance to any of the existing members; for example, if the user has specified a distance of 2, the module will check if the individual is at a distance of 0, 1 or 2 from any existing members. If this requirement is met, the individual is added to the list of potential community members. Next, the individual in the list with the fewest connections at the specified sequence distance (Fig. 2 inset) will be added to the community. Ties for the fewest connections are broken by randomly selecting an individual. The module will continue the process as described until there are no more individuals that meet the requirement for addition to the potential community member list. Current hierarchical clustering algorithms do not guarantee all sequences within a cluster are the specified distance from sequences within another cluster (Westcott & Schloss, 2015), which is essential to DISCo-microbe, motivating us to develop the currently implemented algorithm. Once the community list is complete, the program will output a tab-delimited text file of community members. The community list can be combined with metadata information (optional), such as taxonomic information, which is recommended if the user will be using the ‘subsample by proportions’ option later. A FASTA file of the community list can also be created if desired.

Figure 2: Workflow schematic of the loop that adds new members to the community, starting with the pairwise distance dictionary.
Inset: schematic of adding members with fewest connections at a specified DNA distance. Circles represent individuals, and lines indicate that the connected individuals are at a sequence distance of 3. Green indicates user input of file.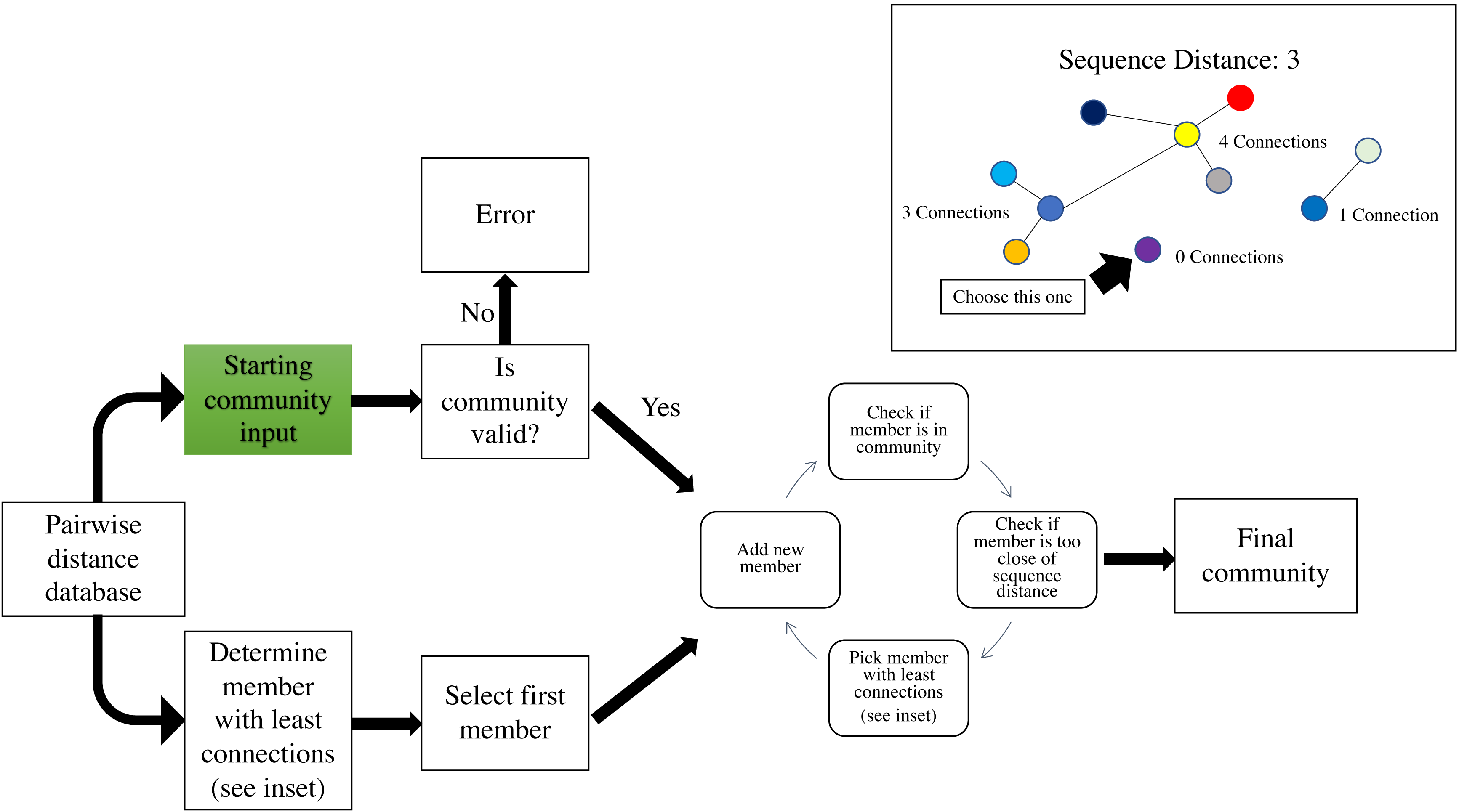{kind=link}
subsample module
The subsample module is designed to take the final output community from the create module and provide a subsample of the community. The module has multiple subsampling procedures. The first method is a random sampling (option: –p-num-taxa) of the indicated number of members, nfinal. The second method (option: –p-proportion) is for subsampling the specific proportions of a grouping variable. To illustrate the use of this option, we will refer to taxonomic information as the grouping variable; however, the user may provide any grouping variable for subsampling. For this option, the user will input two files: the community file from the create module with taxonomic information combined, and a file of the taxonomic groupings with desired proportions. DISCo-microbe will then generate a subsampling of the original community that is optimized to reflect the desired proportions. The optimization is accomplished through a greedy minimization of the sum of differences, , for the set TG of taxonomic groups specified in file 2 (taxonomic proportions file). Here, and are vectors of taxonomic group frequencies for the current and desired community, respectively, with and . The algorithm initializes fcurrent as the vector finput of taxonomic group frequencies of the community provided in file 1 (from create module) with members belonging to taxonomic groups in the set X, where groups not specified in file 2 are removed (X ≡ {x ∈ X|x⁄ ∈ TG}), and finput renormalized such that . Next, the algorithm will continuously iterate the following three steps:
(1) Determine the taxonomic group with largest difference in taxonomic group frequencies, .
(2) If the number of members in the taxonomic group identified in step 1 is less than 2 (ntmax < 2) break and output the current community; otherwise, randomly remove a member from tmax, resulting in fcurrent′.
(3) If , set otherwise stop the module and output the current community.
The user can modify the behavior of the algorithm by specifying both the number of members and the taxonomic proportions (–p-num-taxa and –p-proportion). Providing both options will force the algorithm to continue until the total number of members in the community, ntotal, is ≤nfinal (user-specified final number of members). Further, when both options are specified, step 2 of the greedy minimization is modified to not break iteration when ntmax < 2, and instead removes a member from the taxonomic group with the next-largest difference in frequencies, tnext, where ntnext ≥ 2. Additionally, if the force number option (option: –p-taxa-num-enforce) is used along with –p-num-taxa and –p-proportion, the algorithm will stop iteration when ntotal = nfinal regardless of whether the sum of frequency differences could be further minimized.
Test data set
The Ribosomal Database Project (Cole et al., 2014) file of 16S rRNA genes was downloaded (release 11.5, May 2019), and uncultured strains were using fasgrep (Lawrence et al., 2015). The alignment was trimmed to the V4 region, which is a commonly used region for next-generation sequencing of bacterial communities (Thompson et al., 2017). The initial file contained 239,244 sequences and was randomly subsampled to 10,000 sequences due to the computational intensity of building the community. A reference-based alignment against the SILVA database v. 132 (Pruesse et al., 2007) was created using the program SINA (Pruesse, Peplies & Glöckner, 2012). Alignment sites containing only gaps were removed using alncut (Lawrence et al., 2015). Additionally, 15 sequences aligned poorly and were removed, resulting in a final alignment of 9,985 sequences at a length of 502 bp. The 9,985-sequence alignment was used to create a highly diverse community at a minimum pairwise sequence distance of 3, with the seed set to 10 for reproducibility. Following construction, the subsample module was used to subsample the community to mimic the taxonomic composition a plant-associated microbiome. The final alignment, taxonomic proportion file, and commands used to create the community are available on GitHub for users to reproduce.
Benchmarking
We performed benchmarking on the distance database calculation and the full create command using hyperfine (https://github.com/sharkdp/hyperfine). Benchmarking was performed on a MacBook Air with 1.3 GHz Intel Core i5 with 10 replicate runs per benchmark. To perform the benchmarking, we subsampled the 16S ribosomal test dataset described above using the subsample command, to 50, 100, 250, 500, 1,000, 2,500, 5,000, 7,500, and the full 9,985 sequences for both the distance database calculation and the full create command.
Simulated Illumina data
We simulated 2 × 250 bp paired-end Illumina MiSeq sequencing data for the 16S rRNA RDP community described above using ART v2.5.8 (Huang et al., 2012) with the provided empirical error models for the Illumina MiSeq. We generated three different simulated sequencing data sets with 500 sequences per community member and two samples per simulation. The simulated data was analyzed using two post-processing workflows. The first workflow merges the forward and reverse reads using PEAR (Zhang et al., 2014) followed by dereplicating the sequences using FAST (Lawrence et al., 2015). The second workflow utilizes the dada2 pipeline (Callahan et al., 2016), a program commonly used in the analysis of microbial amplicon sequencing. The dada2 program models Illumina sequencing error and attempts to correct errors to recover the true sequence variants. The resulting sequences of both workflows were assigned to community members using the consensus BLAST (Altschul et al., 1990) method implemented in QIIME2 (Bolyen et al., 2019) with a 99% identity and 99% query length cutoff against the database of community member sequences. Using the community member assignment output, we determined the percent of sequences assigned to community members, percent of community members recovered, and for the dereplicated workflow, the accuracy of community member assignment. Unfortunately, the dada2 pipeline doesn’t provide a mapping of the predicted sequence variants to sequencing reads preventing us from determining the accuracy of community member assignment. Sequences that were unassigned by the consensus BLAST method were searched against the community member sequences using BLASTN keeping the top two hits.
Results
Workflow example
To demonstrate the applicability, usability, and ease of documenting workflows when using DISCo-microbe to construct identifiable diverse communities, we created and subsampled a community with a minimum sequence distance of 3 using 16S rRNA sequences from the RDP database. The initial sequence alignment contained the V4 region from 9,985 sequences with an average pairwise sequence distance of 10.6 ± 3.6%). Using the following create module
command:
disco create –i-alignment RDP_aligned_sequences.fasta –p-editdistance 3 –p-seed 10 –i-metadata
RDP_Metadata_Taxonomy.txt –o-community-list RDP_Community_ED3_seed10.txt
we constructed a community of 2,122 members that could be distinguished through next-generation sequencing. Using the following subsample module
command:
disco subsample –i-input-community RDP_Community_ED3_seed10.txt –p-seed 10 –p-group-by Class –p-proportion RDP_Class_Proportions_file.txt
the community was reduced to 784 community members with the approximate proportions of a plant–associated microbiome (Table 1; Cregger et al., 2018). The options for each module used above, along with the version of DISCo-microbe and Python, are the only documentation required to reproduce the design of this extremely complex community.
| Bacterial class | Input proportions | Output proportions |
|---|---|---|
| Actinobacteria | 0.0885 | 0.0906 |
| Alphaproteobacteria | 0.1857 | 0.1875 |
| Anaerolineae | 0.004 | 0.0013 |
| Aquificae | 0.0003 | 0.0013 |
| Bacteroidia | 0.1 | 0.0982 |
| Betaproteobacteria | 0.1286 | 0.1301 |
| Chitinivibrionia | 0.004 | 0.0013 |
| Chloroflexia | 0.005 | 0.0051 |
| Deferribacteres | 0.0003 | 0.0013 |
| Deinococci | 0.0003 | 0.0026 |
| Deltaproteobacteria | 0.0418 | 0.0434 |
| Fibrobacteria | 0.0004 | 0.0026 |
| Fusobacteriia | 0.0003 | 0.0026 |
| Gammaproteobacteria | 0.4112 | 0.4133 |
| Gemmatimonadetes | 0.0073 | 0.0026 |
| Ktedonobacteria | 0.0097 | 0.0013 |
| Nitrospira | 0.0036 | 0.0051 |
| Planctomycetia | 0.009 | 0.0102 |
Benchmarking
As the number of sequences increased the time to calculate the distance database and to create the full community increased exponentially (Fig. 3). Upon examination, the distance database was the most computationally expensive portion of the create module responsible for between 55 and 95% of the total time to create the community (Fig. 3). The full community construction with the alignment of 9,985 sequences using the create module took on average 13.09 min (±4.42 s) with 12.26 min (±4.01 s) being the distance database calculations.
Figure 3: Benchmarking of distance database and create module.
Benchmarking of custom nucleotide Hamming distance function for DNA and the create module at various numbers of 16S rRNA sequences subsampled from the Ribosomal Database Project dataset.{kind=link}
DISCo-microbe designs communities with members distinguishable by amplicon sequencing
We simulated Illumina MiSeq sequencing data from the 2,120 member community constructed from the 9,985 16S rRNA sequences from the RDP database and described above. Unexpectedly, sequencing data was only generated from the first 2,065 community members due to an undocumented limit on the number of input sequences that ART (Huang et al., 2012) will process, however this does not change the overall results of the analysis. We noticed that ART simulated sequencing data consistent with empirically determined error rate of 0.24% errors per base (Pfeiffer et al., 2018). However, an average of 25% of the simulated sequences contained an error compared to an average of 6.4% of empirical sequences (Pfeiffer et al., 2018). Using the dereplication workflow, we were able to recover sequences from all 2,065 community members (Fig. 4A) and 97.7% (±0.0004%) of dereplicated sequences were assigned to a community member with the remaining 2.3% of sequences unassigned (Fig. 4B). Notably, none of the sequences were misclassified. Using the dada2 workflow, we recovered sequences from fewer of the community members (97.8% ± 0.0007%) compared to the dereplication workflow (Fig. 4A) but had a higher rate (99.3% ± 0.001) of sequence variants assigned to a community member (Fig. 4B). BLASTing unassigned sequences against the community member sequences mostly resulted in the top hit being the correct community member. Unexpectedly, one of the unassigned sequences from the dada2 workflow only had one nucleotide different from two community members. Upon further examination of these two community members we identified an alignment error in the alignment used to create the community that when corrected resulted in the two community members having a pairwise distance of 2 instead of the required 3.
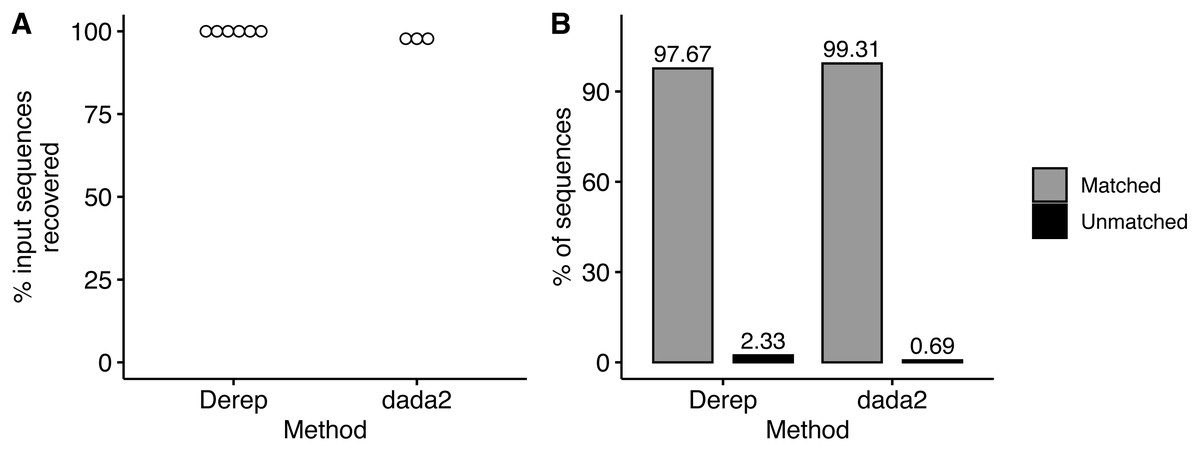
Figure 4: Recovery and taxonomic assignment of sequences from Illumina simulator.
(A) The percent of sequences from the input community that were recovered from the Illumina simulator with the dereplication and the dada2 method. (B) The percent of sequences that had a taxonomic assignment or were not assigned.{kind=link}
Discussion
Microbial diversity is linked to function (Turnbaugh et al., 2008; Laforest-Lapointe et al., 2017), but understanding that diversity can be difficult due to the low resolution of taxonomic marker genes and the complexity of the microbial community, limiting our ability to identify and track individual community members. To tease apart the complex interactions within communities, there has been an increased demand for synthetic community systems (Busby et al., 2017). However, the generation of complex communities of organisms that can be easily distinguished through high-throughput methods can be difficult without strong computational skills. In general, two challenges are associated with the design of a synthetic community: (1) creation of a distinguishable community through common sequencing methods and (2) development of a community with the desired traits. Additionally, manual creation can lead to a lack of reproducibility due to the difficulty of documenting the workflow. In this paper, we describe an easy to use command-line program, Design of an Identifiable Synthetic Community of Microbes (DISCo-microbe), for the creation of diverse communities of organisms that can be distinguished through next-generation sequencing technology during in vivo experiments. DISCo-microbe solves the two previously mentioned problems using two modules, create and subsample.
The create module allows the user to construct a diverse community that is identifiable using common sequencing methods, thus solving the first problem. The ability to specify a minimum sequence distance allows flexibility in the construction of the community due to its robustness to sequencing errors introduced through PCR and sequencing (Pfeiffer et al., 2018). For example, if the user sets the minimum sequence distance to 5, sequences containing up to 2 sequencing errors ([d − 1]∕2) can be confidently assigned to the correct community member, sequences containing up to 4 errors (d − 1) can be identified, and it would take a minimum of 5 errors to assign a sequence to the incorrect community member. Usually, the smaller the minimum sequence distance, the more members will be included in the constructed community, potentially motivating users to set the minimum sequence distance to lowest setting of 1. However, at a minimum sequence distance of l, it only requires a single sequencing error to assign a sequence to the wrong community member. In order to implement the create module, we developed a custom nucleotide Hamming distance that accommodates nucleotide ambiguities. This is the first application of the Hamming distance algorithm incorporating IUPAC nucleotide ambiguity codes to measure distance between pairs of aligned sequences implemented in Python (see Šošić & Šikić, 2017 for an implementation in C). We determined that the most time-consuming step is the creation of the distance database due to the number of calculations required . Despite the large number of calculations required to create the distance database, the runtime for the create module on the largest community containing 9,985 sequences was only 13 min on a MacBook Air laptop.
The subsample module allows flexibility in the final constructed community. Specifically, it allows users to adapt the community to their experimental specifications, either by limiting the number of strains, specifying proportions of a grouping variable, or both. The subsample module eliminates major problem (2) by allowing users to tailor the already distinguishable community to include desired traits or proportions of members, examples of which are found in the detailed documentation.
Using simulated Illumina MiSeq data, we demonstrated the ability of DISCo-microbe to design diverse communities with members distinguishable by amplicon sequencing. We were able to identify sequences from 97.5% and 100% of community members when using the dada2 and dereplication workflows respectively. Notably, when using the dereplication workflow, we show that we do not have any misclassified sequences indicating that all members were distinguishable. Furthermore, the inability to assign 2.3% and 0.7% of sequences to community members in the dereplicate and dada2 workflows respectively were a result of multiple sequencing errors. The number of unassignable sequences in our simulated data is likely an overestimation compared to real data. Given that 25% of ART simulated Illumnia MiSeq reads had at least one error compared to the recently documented empirical rate of 6% (Pfeiffer et al., 2018). Despite the greater number of sequences being mutated than expected in a real sequencing run, we still show the ability to discriminate between community members with a high degree of accuracy and recall. Further investigation into the unassigned sequences using BLASTN demonstrated the ability to accurately assign all but one of these sequences based on their top BLAST hit against the community member sequences. Consequently, increasing the overall percent of sequences assigned to community members and percent of community members recovered without increasing our false positive rate. The only sequence unassignable by BLASTN was a dada2 sequence variant that only has a single nucleotide difference from two community members. Upon further investigation of these two community members we discovered errors in the alignment resulting in an overestimation of the distance between these two community members. This illustrates the dependence of DISCo-microbe on an accurate input alignment to determine the correct distance between individuals, and thus creating a community at the desired sequence distance. Notably, despite this alignment error the dereplication workflow along with BLASTN was able to accurately distinguish all community members making the community still identifiable.
Conclusions
DISCo-microbe is the first software designed for the construction of a diverse community of organisms that can be distinguished through low-cost, high-throughput amplicon sequencing for use in in vivo experiments. DISCo-microbe allows non-programmers to easily and reproducibly construct communities in which the members are identifiable through amplicon sequencing and the communities conform to user-specified attributes or numbers of members. DISCo-microbe is also the first software to implement a nucleotide specific Hamming distance in Python that takes into account nucleotide ambiguities in sequencing data. Although initially designed for bacterial community construction, the input of a nucleotide sequence alignment from any region allows the software to be used with any group of organisms. DISCo-microbe is designed for easy expansion of utilities; planned future versions will include new algorithms for community construction as well as new modules for creating a suite of tools for the design of constructed communities and processing of the resulting data.
Availability and requirements
Project name: DISCo-microbe
Project home page: https://github.com/dlcarper/DISCo-microbe
Operating system(s): platform-independent
Programming language: Python ≥ 3.4
Other requirements: BioPython
License: GNU General Public License v3.0