
Adsorption behavior of CO2 molecule on AlN and silicene—application to gas capture devices
- Published
- Accepted
- Received
- Academic Editor
- Junkuo Gao
- Subject Areas
- Graphenes and Fullerenes, Nano and Microstructured Materials
- Keywords
- Gas capture, CO2 adsorption
- Copyright
- © 2020 Jia and Luo
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Materials Science) and either DOI or URL of the article must be cited.
- Cite this article
- 2020. Adsorption behavior of CO2 molecule on AlN and silicene—application to gas capture devices. PeerJ Materials Science 2:e3 https://doi.org/10.7717/peerj-matsci.3
Abstract
Carbon dioxide contributes significantly to both global warming and climate change, processes that inflict major environmental damage, which is why it is of much interest to find a material that can adsorb carbon dioxide before it enters the atmosphere. In our study, we use first-principles calculations based on the density functional theory to investigate the adsorption of carbon dioxide on two-dimensional materials due to their unique chemical and physical properties. The two-dimensional materials we used include aluminum nitride, defected aluminum nitride, and silicene. We observed a negative adsorption energy of carbon dioxide on all three materials, signifying a spontaneous adsorption. Our charge analysis reveals a charge transfer from the materials to the molecule in addition to a significant overlap between the projected density of states spectra of the interacting atoms, all indicating the formation of chemical bonds between the material and adsorbed molecule. Our findings thus suggest that all the materials we used could be an effective adsorbent for carbon dioxide; however, the defected aluminum nitride sheet formed stronger bonds with carbon dioxide compared to the pure sheet. The application of our research could help decrease the world’s carbon footprint by creating devices to capture carbon dioxide before it enters the atmosphere.
Introduction
Carbon dioxide is a greenhouse gas, a type of gas that adsorbs heat and gradually releases it over time (Lashof & Ahuja, 1990). It accounts for around 86% of man-made greenhouse gases, and greatly factors into environmental issues such as climate change and global warming (Solomon et al., 2009). In 2013, the global average parts per million (ppm) of carbon dioxide was 400 ppm, the highest it has ever been in the past 600,00 years (Berner, 1998). Over the past 60 years, the rate of increase of carbon dioxide in the air has grown from 0.6 ppm in the 1960s to 2.3 ppm today, and scientists predict that this number will only increase (Taub, 2010). With an increase of carbon dioxide comes rising sea levels due to the melting of ice and glaciers, lower food production because of rising temperatures, and extreme precipitation due to the intensification of Earth’s water cycle (Lemoine, 2018). The problem with rising carbon dioxide levels is further amplified by the human destruction of natural carbon dioxide sinks (Hampicke, 1979). A carbon dioxide sink is a natural reservoir that stores carbon dioxide over an indefinite period of time (Sarmiento & Gruber, 2002). The main natural sinks for carbon dioxide include forests, which have continuously shrunk due to deforestation (Le Quéré et al., 2009; Sarmiento & Gruber, 2002). Because of the increasing concern and destructive impacts of increasing carbon dioxide levels, it is imperative to find a material that can adsorb carbon dioxide in order to relieve the problems of climate change and global warming. For the past 50 years, post-combustion ‘wet-scrubbing’ CO2 capture technology has been employed to help decrease the amount of carbon dioxide leaving industrial plants (Bosoaga, Masek & Oakey, 2009). This process refers to capturing carbon dioxide from the gas after the burning of fossil fuels (Merkel et al., 2010). However, this method requires considerable amounts of energy, around 4 trillion kilowatt hours, in order to operate properly (Bhown & Freeman, 2011). In addition, the solvents used during this process are prone to chemical degradation which leads to reduced efficiency and increased costs to help maintain this process (D’Alessandro, Smit & Long, 2010). Previous studies using nanocrystals and nanotubes have been shown to identify new materials that can be used to adsorb carbon dioxide (Wang, Pennycook & Pantelides, 2002).
Studies using cadmium selenide nanocrystals have revealed that carbon dioxide molecules are able to bond to defected surfaces with a deficiency of Se atoms. The carbon dioxide molecules then become negatively charged and more reactive with organic materials (Gupta, Sakthivel & Seal, 2015). Previous literature studying the adsorption of carbon dioxide molecule has mainly focused on using nanocrystals and nanotubes (Hussain et al., 2018). However, 2D monolayers such as graphene exhibit unique electrical properties and have extremely thin and lightweight structures compared to already studied nanocrystals and nanotubes (Neto et al., 2009). This is what prompted us to research carbon dioxide adsorption on 2D monolayers, specifically aluminum nitride and silicene.
Previous studies using a group III nitride, boron nitride, have shown that boron nitride can effectively adsorb carbon dioxide, leading us to research the adsorption capabilities of other group III nitrides (Jiao et al., 2011). Aluminum nitride is a group III nitride with a flat hexagonal structure (Feng et al., 2019). It was recently synthesized in 2015 and is of much interest due to its large band gap and semiconductor nature which allows for it to be used in many potential devices (Şahin et al., 2009). Aluminum nitride nanotubes have been studied extensively as an adsorbent for carbon dioxide. However, the nanotubes are hard to synthesize and only nanotubes with a small enough diameter were found capable of adsorbing carbon dioxide (Pinhal et al., 2019; Jiao et al., 2011). In addition, we implemented a nitrogen vacancy to conclude whether or not defected sheets are better for adsorbing materials. Unlike aluminum nitride, silicene is not a flat monolayer and contains a buckling height structure with Si atoms both below and above the 2D structure. These regions have distinct electronic charges making silicene more reactive than the other two materials (Chen et al., 2016). Past studies have shown that buckled monolayers are able to adsorb carbon dioxide (Deshpande et al., 2019). We chose silicene to determine whether the shape of the material impacts the adsorption capability.
This study aims to implement first-principles investigation using the density functional theory to investigate the interactions between different 2D monolayers and their adsorption of carbon dioxide (Gonze et al., 2002). Analysis of these effects are vital towards the mitigation of climate change and its effects such as rising global temperatures. In this study, we report the specific first-principles plane-wave basis computational methods used, results, and direct interpretation of the calculations. We also discuss our findings as well as the future implications for this research in order to supplement our findings.
In ‘Methods’, we detail our methods to perform first-principles calculations. In ‘Results’, we present our results on carbon dioxide and our monolayer configurations. In ‘Discussion’, we discuss and compare our results with experimental and other theoretical research. Finally, our conclusion and future work are found in the ‘Conclusion’ section.
Methods
We performed first-principles calculations based on the density functional theory (DFT) in the ABINIT code (Gonze et al., 2009). We used the Generalized Gradient Approximation (GGA) in Perdew-Burke-Erzenhof (PBE) form as our exchange correlation functional. We used the Projected Augmented Wave (PAW) (Blöchl, 1994) pseudopotential method with projectors generated using the Atom-PAW code (Holzwarth, Tackett & Matthews, 2001).
Convergence calculations were performed to find the converged kinetic energy cutoff, k-point mesh, and vacuum height. The values were considered converged when the total energy difference between datasets was less than 1.0 × 10−4 Ha twice consecutively. Each self- consistent field (SCF) iteration was terminated when the difference between the total energy was 1.0 × 10−10 Ha. In addition, we relaxed the lattice parameters using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm to determine the optimized lattice parameters. Relaxation calculations were completed when the maximum forces converged to less than 5.0 × 10−4 Ha/Bohr. The 2D materials we used for our monolayers include aluminum nitride, and silicene. We performed convergence calculations on a 1 × 1 unit cell of our monolayers. Using the converged values, we then found the optimized lattice parameters using full relaxation. We also ran convergence calculations for a carbon dioxide molecule suspended along the horizontal axis of an empty box. We used one k-point for the isolated carbon dioxide system, and found a kinetic energy cutoff of 20 Ha. The carbon dioxide molecule was placed on top of a 2 × 2 supercell of our monolayer. When combining the surface with the carbon dioxide molecules, we used the higher energy cutoff value.
The adsorption energy for each of our configurations is defined by: (1) where Emol+surf, Esurf, and Emol represent the total energies of the interfaces, the monolayer surface, and isolated carbon dioxide molecule respectively. From the total energy output data, we visualized charge transfer distributions in bonded interfaces. Charge transfer is defined by: (2) where ρmol∕surf(τ), ρsurf(τ), and ρmol(τ) represent the total charge of the carbon dioxide and monolayer interfaces, monolayer surface, and isolated carbon dioxide molecule respectively. Charge transfer distributions were plotted in XCrysDen.
In addition, we also plotted the projected density of states (PDOS) for our pristine monolayers and complexes. We looked for strong hybridization and overlapping orbitals to indicate the formation of chemical bonds between the carbon dioxide molecule and monolayer. One limitation of using GGA-PBE functional is that it tends to underestimate the band gap. By using a 2 × 2 supercell of our monolayers, we increased the size of our monolayer. Since carbon dioxide is a relatively smaller molecule, we believed that there would be minimal self-interaction because of the size difference. This would make it so that the band gap can be made close to experimental or high-level theoretical prediction.
Results
First we studied the optimized structures of the carbon dioxide molecule and the three monolayers. Next, we calculated the adsorption energy of carbon dioxide on our monolayers. We wanted to find an adsorption energy of greater than 0.3 eV or approximately 0.011 Ha to indicate a chemisorption of carbon dioxide onto the sheet. Electronic structures were further analyzed through plotting both the PDOS and charge transfer.
Pure carbon dioxide molecule and surfaces
We compared the lattice parameters of our surface and bond lengths of carbon dioxide with experimental data in Table 1.
| C = O Bond Length | AlN (a) | Silicene (a) | |
|---|---|---|---|
| Calculated (Bohr) | 2.214 | 5.920 | 7.285 |
| Theoretical (Bohr) | 2.196 (Jiao et al., 2011) | 5.877 (Feng et al., 2019) | 7.266 (Feng et al., 2014) |
| Experimental (Bohr) | 2.192 (Taylor, 1983) | 5.914 (Bacaksiz et al., 2015) | 7.313 (Drummond, Zolyomi & Fal’Ko, 2012) |
| Error (%) | 0.993 | 0.103 | 0.384 |
Since silicene has a buckled structure, it has an additional constant for the buckling height. We calculated the buckling height of silicene and found it to be 0.827 Bohr with a 0.5% error (Shobha, Pratap & Sumit, 2015).
Carbon dioxide on pure aluminum nitride
Atomic structure
To find the most favorable binding sites of carbon dioxide molecules on the aluminum nitride monolayer, we considered two possible adsorption sites. We tested two different configurations of carbon dioxide over a pure aluminum nitride sheet, placing the carbon dioxide molecule parallel to the sheet over both the aluminum and nitrogen atom (Fig. 1). We ran full relaxation for each of our configurations and used the relaxed values to determine the adsorption energy of the complex.
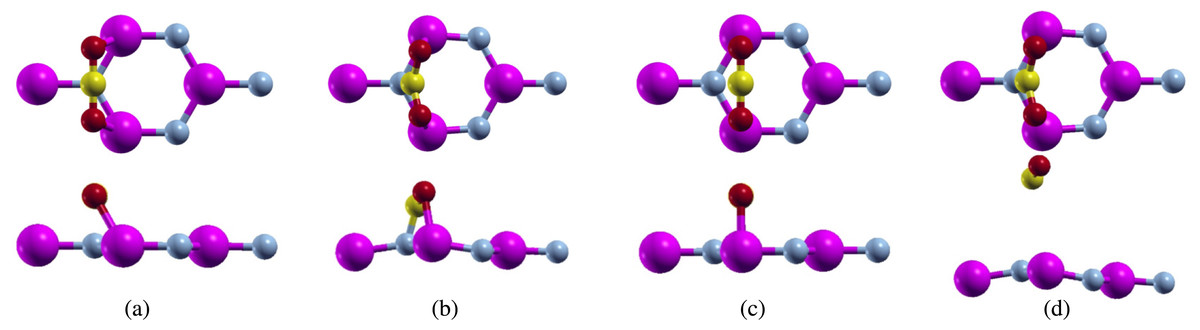
Figure 1: Top and side view of carbon dioxide on pure aluminum nitride (purple atoms are aluminum, blue atoms are nitrogen, yellow atoms are carbon, and red atoms are oxygen) positioned parallel to the N atom (A) before relaxation (B) after relaxation, and positioned parallel to the Al atom (C) before relaxation (D) after relaxation.
.{kind=link}
For the first configuration, we proposed the bonding of the carbon atom from carbon dioxide over the nitrogen atom in the aluminum nitride sheet with the carbon dioxide molecule laying parallel to the monolayer (Fig. 1A). Although we placed the carbon dioxide molecule over the nitrogen atom, it formed bonds with the aluminum atoms instead. The calculations were carried out using a full relaxation method meaning any attractions formed were stable chemical bonds.
After relaxation, we noticed that the configurations had changed (Fig. 1B). We measured the C-O bond length and the O-C-O bond angle. Before relaxation, the C-O bond length was 2.214 Bohr, while the O-C-O bond angle was 180°. We noticed a lengthening of the C-O bond length to 2.405 Bohr while the O-C-O bond angle shrunk to 128.747°. We can attribute the lengthening of the C-O bond length to the π bonds breaking when this configuration relaxed.
Next, we proposed the bonding of the oxygen atoms from carbon dioxide to the aluminum atoms in aluminum nitride (Fig. 1C). After running full relaxation on this configuration, we noticed that the carbon dioxide molecule had moved away from the aluminum nitride sheet, leading us to believe that the aluminum atoms are not a viable adsorption site for carbon dioxide (Fig. 1D). Further calculations, adsorption energy, PDOS, and charge transfer, were not calculated for this adsorption site.
Adsorption energy
For the equation we used, a negative adsorption energy indicated an exothermic reaction, meaning the configuration resulted in a spontaneous adsorption. On the other hand, a positive adsorption energy indicated an endothermic reaction, resulting in a non-spontaneous adsorption of carbon dioxide. In addition, the larger the magnitude of the adsorption energy, the more stable the configuration is. We tried to find configurations that have a large negative adsorption energy, indicating a stable spontaneous adsorption. Both the sign and magnitude had to be taken into account when determining whether or not the configurations were likely to adsorb.
The adsorption energy that we calculated for the bonding of carbon dioxide over the nitrogen atom was −0.027 Ha. The negative value we observed suggests a probable spontaneous adsorption of the carbon dioxide molecule onto aluminum nitride over the nitrogen atom. In the adsorption of carbon dioxide over aluminum nitride, the magnitude of the adsorption energy, 0.027 Ha, is greater than 0.011 Ha meaning that carbon dioxide is chemisorbed onto aluminum nitride. Since the carbon dioxide molecule did not show a binding affinity when bonded over the aluminum atom, we can conclude that the nitrogen atom is the better binding site for a pure aluminum nitride sheet.
Projected density of states
To further discover the mechanism of chemisorption of carbon dioxide molecules onto our monolayers, we investigated the electronic structures of these configurations by plotting the projected density of states as well as the charge transfer. For the PDOS, we plotted the 2p orbitals of both oxygen and carbon from the carbon dioxide molecule in addition to the valence orbitals of the elements that make up our monolayers.
For the adsorption of carbon dioxide on aluminum nitride, we plotted the 3p orbitals of aluminum and 2p orbitals of nitrogen in addition to the aforementioned carbon dioxide orbitals (Fig. 2B). In addition, we plotted the PDOS for a pure aluminum nitride sheet (Fig. 2A). It can be seen from the PDOS of carbon dioxide on aluminum nitride that there is a significant overlap between the PDOS spectra of the interacting aluminum, nitrogen, and oxygen atoms, indicating the formation of chemical bonds between them.
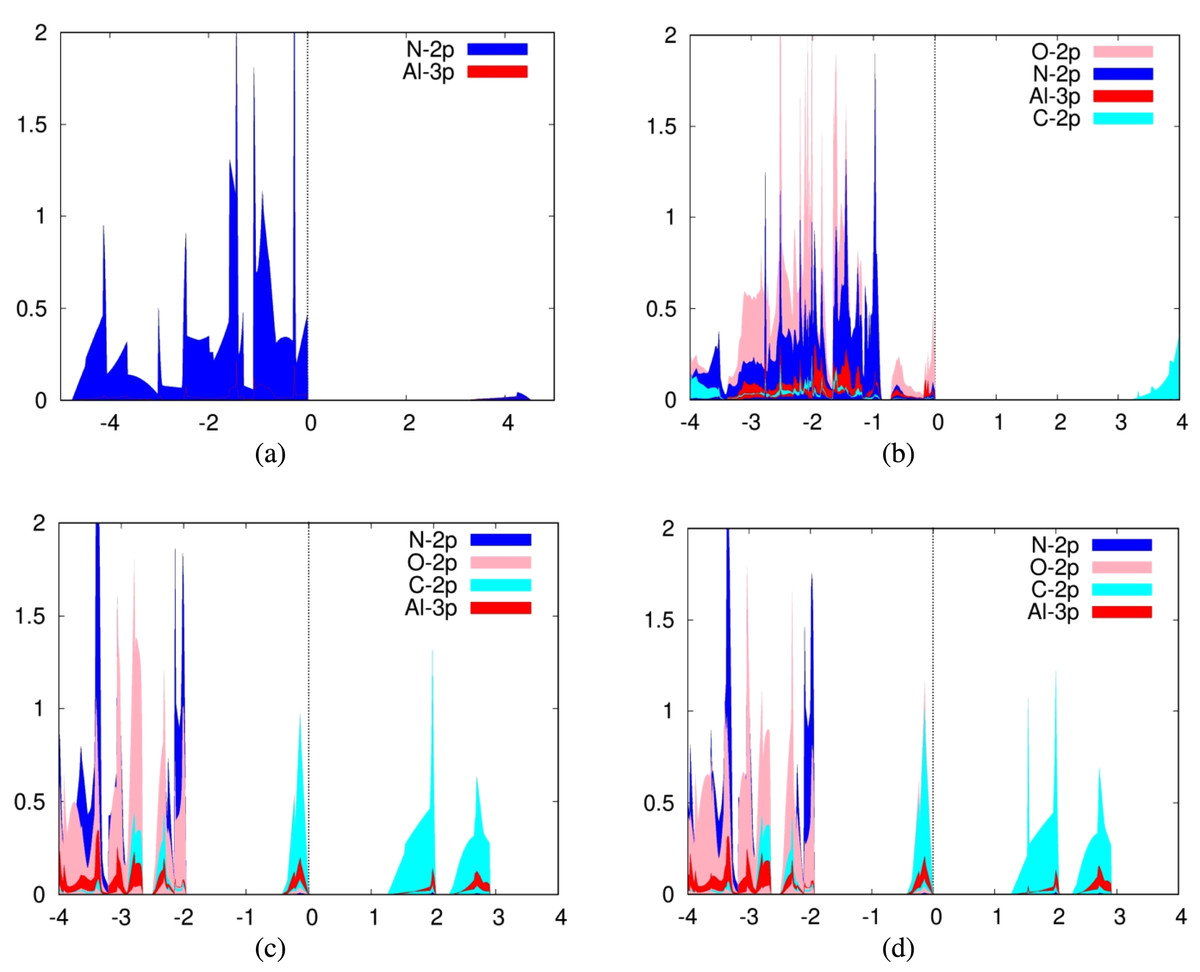
Figure 2: Projected density of states graph for (A) pure aluminum nitride sheet (B) CO2 placed parallel to the N atom over aluminum nitride (C) CO2 placed parallel to the N atom over defected aluminum nitride (D) CO2 placed parallel to the Al atom over defected aluminum nitride.
{kind=link}
Charge transfer
The formation of chemical bonds was also confirmed by the transfer of electrons between the carbon dioxide molecule and aluminum nitride sheet as shown in the charge transfer (Fig. 3). When we plotted the charge transfer, we observed that the aluminum atoms lost charge, while both the nitrogen atoms and oxygen atoms in carbon dioxide gained charge. Oxygen is more electronegative than both aluminum and nitrogen (1.61 for aluminum and 3.04 for nitrogen vs. 3.44 for oxygen) because oxygen only needs two more electrons to fill its valence shell, while aluminum needs to lose three electrons and nitrogen needs to gain three electrons to achieve a full valence shell. The presence of charge transfer signifies that bonding did in fact occur between the carbon dioxide molecule and aluminum atoms.

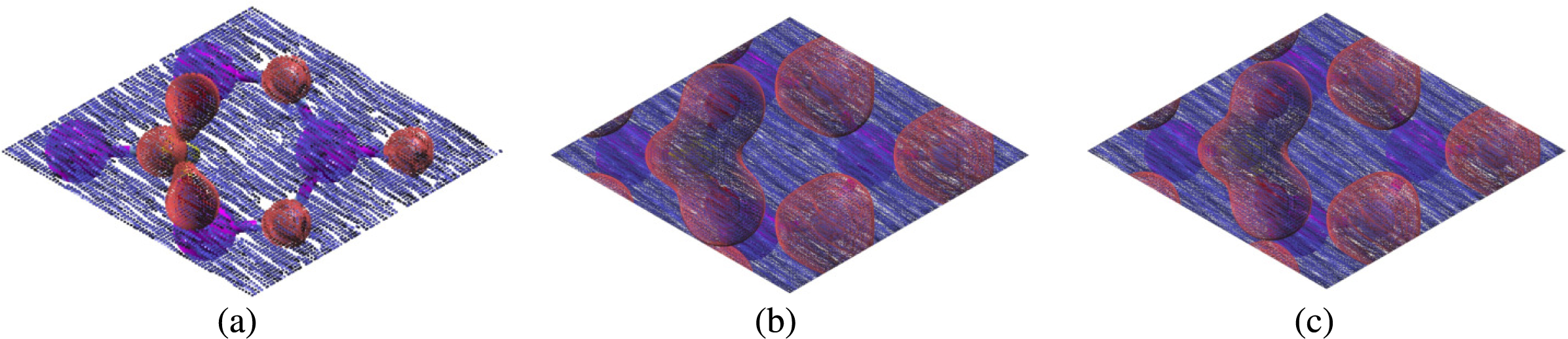
Figure 3: Charge transfer of carbon dioxide molecules on monolayer (purple atoms are aluminum, blue atoms are nitrogen, yellow atoms are carbon, red atoms are oxygen; charge transfer plots: blue is charge lost, red is charge gained) the isosurface value we used was 0.3 e/Bohr3 (A) CO2 parallel to the nitrogen atom on aluminum nitride (B) CO2 parallel to the nitrogen atom on defected aluminum nitride (C) CO2 parallel to the aluminum atom on defected aluminum nitride.
{kind=link}
Carbon dioxide on defected aluminum nitride
Atomic structure
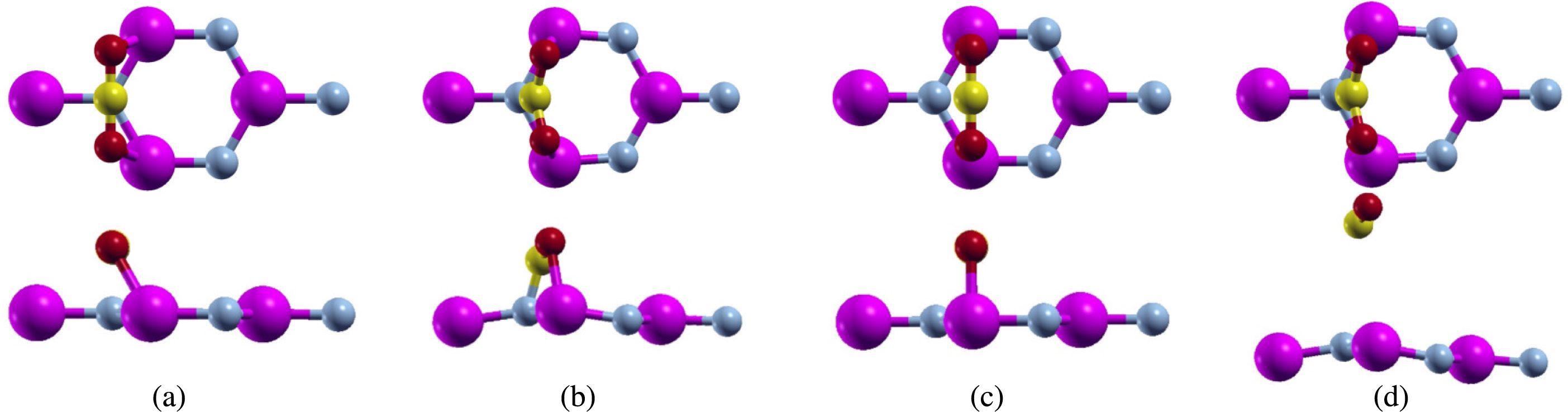
Since pure aluminum nitride wasn’t able to adsorb carbon dioxide over both adsorption sites, we tried to make the sheet more reactive by implementing a single atom vacancy. To create the vacancy, we removed the nitrogen atom over which the carbon dioxide was bonded over. Again, we proposed the same configurations of carbon dioxide on top of the sheet as the pure aluminum nitride (Fig. 4). We ran full relaxation for each of our configurations.
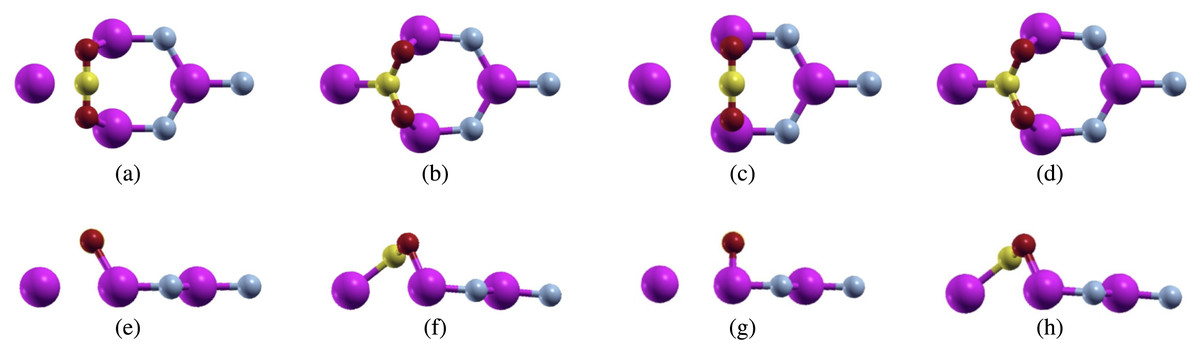
Figure 4: Carbon dioxide on defected aluminum nitride (purple atoms are aluminum, blue atoms are nitrogen, yellow atoms are carbon, and red atoms are oxygen) positioned parallel to the N atom top view (A) before relaxation (B) after relaxation, and positioned parallel to the Al atom top view (C) before relaxation (D) after relaxation, and positioned parallel to the N atom side view (E) before relaxation (F) after relaxation, and positioned parallel to the Al atom side view (G) before relaxation (H) after relaxation.
{kind=link}
For the first configuration, we proposed the bonding of the carbon atom from carbon dioxide over the nitrogen atom. We actually removed this nitrogen atom in order to create a vacancy within the aluminum nitride sheet. However, even though the carbon atom was placed over the nitrogen atom, the carbon dioxide molecule formed bonds with the aluminum atoms, similar to this configuration over pure aluminum nitride (Fig. 4A). The calculations were performed using a full relaxation method. After relaxation, we noticed that the configuration had changed (Fig. 4B). We measured both the C-O bond length and the O-C-O bond angle. Before relaxation, the C-O bond length was 2.214 Bohr, while the O-C-O bond angles was 180°. For the C-O bond length, we noticed that it lengthened to 2.491 Bohr while the O-C-O bond angle shrunk to 118.096°. Although this configuration did change after relaxation, there were no major structural changes as the configuration was still intact.
Next, we proposed the bonding of the oxygen atoms from carbon dioxide to the aluminum atoms in aluminum nitride (Fig. 4C). After running full relaxation on this configuration, we found that the carbon dioxide molecule did bond to the defected aluminum nitride sheet unlike the pure aluminum nitride. This led us to believe that creating single atom vacancies is an effective way to make a sheet more reactive, and thus more likely to adsorb carbon dioxide.
We observed this configuration after relaxation and noticed that it had changed from the original configuration (Fig. 4D). Similar to the previous configuration, we measured the relaxed C-O bond length and O-C-O bond angle. After relaxation, the C-O bond lengthened to 2.492 Bohr and the O-C-O bond angle shrunk to 118.088°. These numbers are quite close to the previous configurations leading us to believe that while both configurations started off in different positions, they both relaxed to a similar position.
Adsorption energy
For a defected sheet of aluminum nitride, we found that the adsorption energy of carbon dioxide over both of the adsorption sites were the same, with an adsorption energy of −0.072 Ha. The fact that both configurations after relaxation have the same adsorption energy further supports the idea that both configurations relaxed to the same or a very similar position.
The magnitude of the adsorption energy of carbon dioxide over defected aluminum nitride is greater than that of carbon dioxide over pure aluminum nitride, meaning that carbon dioxide forms a more stable adsorption with defected aluminum nitride. While carbon dioxide can be chemisorbed onto pure aluminum nitride, a defected sheet of aluminum nitride is better due to the more stable formation.
PDOS
For the adsorption of carbon dioxide on defected aluminum nitride, we plotted the 3p orbitals of aluminum and 2p orbitals of nitrogen in addition to the 2p orbitals of carbon and oxygen (Fig. 2). It can be seen from the PDOS of carbon dioxide on aluminum nitride that there is a significant overlap and hybridization between the PDOS spectra of the interacting aluminum, nitrogen, and oxygen atoms, indicating the formation of chemical bonds between them.
Charge transfer
The formation of chemical bonds was also confirmed by the transfer of electrons between the carbon dioxide molecule and aluminum nitride sheet as shown in the charge transfer (Fig. 3). When we plotted the charge transfer, we observed that the aluminum atoms lost charge, while both the nitrogen atoms and oxygen atoms in carbon dioxide gained charge (Fig. 3). Oxygen is more electronegative than both aluminum and nitrogen (1.61 for aluminum and 3.04 for nitrogen vs. 3.44 for oxygen) because oxygen only needs two more electrons to fill its valence shell, while aluminum need to lose three electrons and nitrogen needs to gain three electrons to achieve a full valence shell. The presence of charge transfer signifies that bonding did in fact occur between the carbon dioxide molecule and aluminum atoms.
Carbon dioxide on pure silicene
Atomic structure
Unlike aluminum nitride, silicene forms a hexagonal buckled lattice. We used low-buckled siliecene with a buckling height of 0.857 Bohr. Silicene is not fully planar because the π electrons are more active (Lian & Ni, 2013), causing a buckled structure to be more stable than a planar structure.
Due to the buckled structure of silicene, there are distinct Siup and Sidn positions. To find the most favorable binding sites of carbon dioxide molecules on a pure silicene monolayer, we considered two possible adsorption sites. For our two configurations, we proposed the bonding of the two oxygen atoms from carbon dioxide to the Siup and Sidn atoms, placing the carbon dioxide molecule parallel to the sheet (Fig. 5). We ran full relaxation for each of our configurations and used the relaxed values to determine the adsorption energy of the complex.
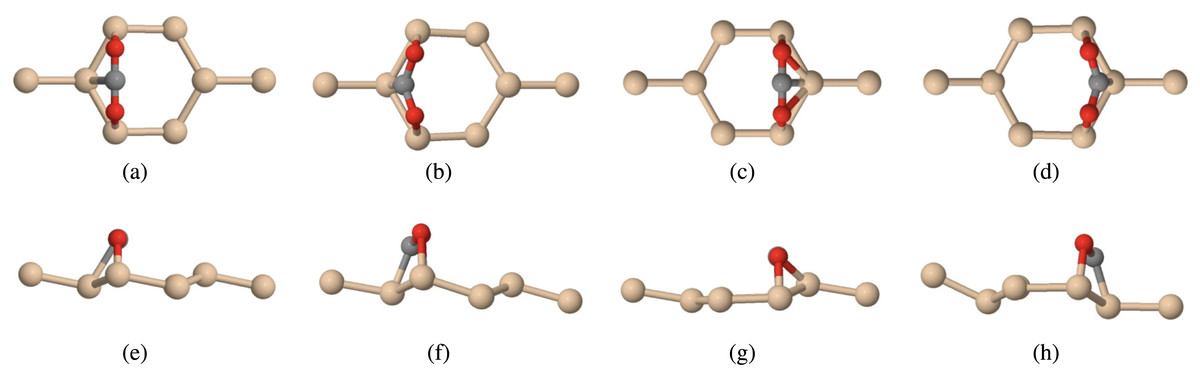
Figure 5: Carbon dioxide on pure silicene (tan atoms are silicon, grey atoms are carbon, red atoms are oxygen) positioned parallel to the Siup atom top view (A) before relaxation (B) after relaxation, and positioned parallel to the Sidn atom top view (C) before relaxation (D) after relaxation, and positioned parallel to the Siup atom side view (E) before relaxation (F) after relaxation, and positioned parallel to the Sidn atom side view (G) before relaxation (H) after relaxation.
{kind=link}
Our first configuration proposed the bonding of the two oxygen atoms from carbon dioxide to Siup atoms (Fig. 5A). This figure shows both the top and side view of our configuration before relaxation. We then ran full relaxation calculations on this configuration to find the optimized structure (Fig. 5B).
Similarly, we also proposed the bonding of the two oxygen atoms from carbon dioxide to the two Sidn atoms (Fig. 5C). As with the first configuration, we used a full relaxation method. After relaxation, we observed that the Sidn atoms moved, making them appear to be Siup atoms (Fig. 5D). This was unusual because Siup atoms are more likely to bond with electropositive elements, while Sidn atoms are more likely to bond with electronegative elements such as oxygen. However, this theory was based off of research done on high-buckled silicene while we used low-buckled silicene. This led us to believe that oxygen atoms are more likely to bind with Siup in low-buckled silicene.
For both of our configurations, we measured the C-O bond length and O-C-O bond angle before and after relaxation. When focusing on the C-O bond length, we noticed that the bond lengthened from 2.214 Bohr to 2.257 Bohr and 2.448 Bohr for the Siup and Sidn configurations respectively. We can attribute the lengthening of the C-O bond to the breaking of the π bonds when both of these configurations relaxed. Furthermore, we noticed that the O-C-O bond angle shrunk from 180°to 122.834°and 122.064°for the Siup and Sidn configurations respectively.
Adsorption energy
For the silicene sheet, we calculated adsorption energies of −0.075 and −0.025 Ha for the adsorption of carbon dioxide over the Siup and Sidn atoms respectively. Both of these values are negative indicating a probable spontaneous adsorption of carbon dioxide onto the silicene sheet and greater than 0.011 Ha indicating a possible chemisorption. However, the magnitude of the adsorption energy when carbon dioxide is bonded to the Siup atoms is greather than the magnitude of the adsorption energy when carbon dioxide is bonded to the Sidn atoms. These magnitudes show that the best adsorption site for carbon dioxide on silicene is over the Siup atoms because it forms a more stable configuration. This is in agreement with our previous atomic structure that also suggests that carbon dioxide molecules are more likely to bond with Siup atoms.
Projected density of states
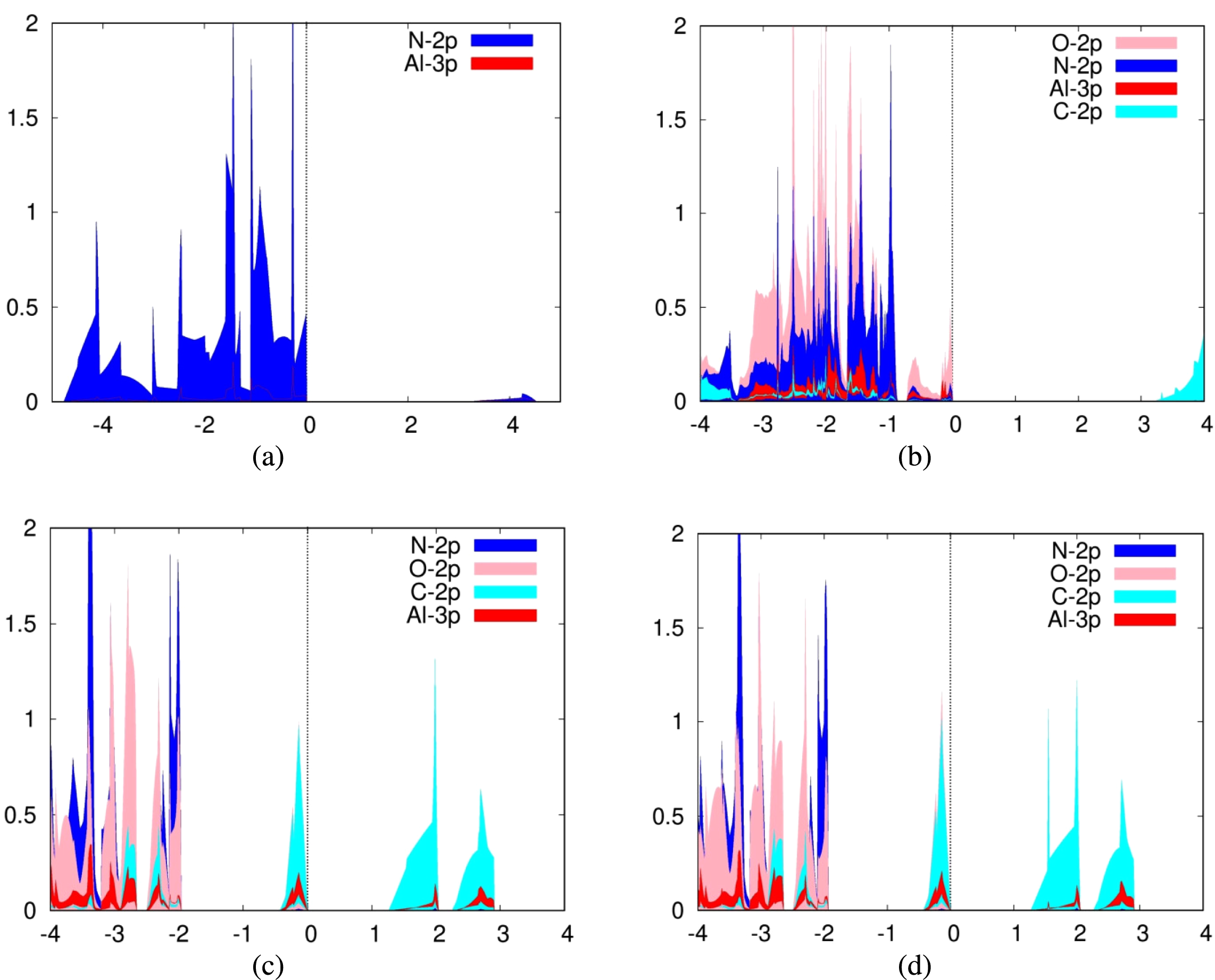
For the adsorption of carbon dioxide on silicene, we plotted the 3p orbitals of silicon in addition to the previously mentioned orbitals of carbon dioxide for the PDOS. Again, we also plotted the PDOS of a pure silicene sheet (Fig. 6A). It can be seen from the PDOS of both carbon dioxide adsorbed over Siup atoms (Fig. 6B) and Sidn atoms (Fig. 6C) that there is a significant overlap between the PDOS spectra of the interacting silicon and oxygen atoms, indicating the formation of chemical bonds between them. Silicene is a good adsorbent for carbon dioxide because the 2p orbitals of the carbon and oxygen atoms are strongly hybridized with the 3p orbitals of silicene. The PDOS for these orbitals fall and rise at the same time.
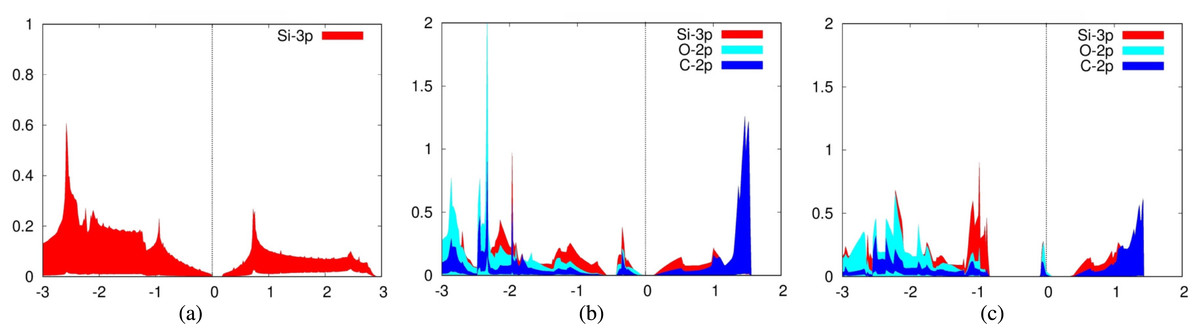
Figure 6: Projected density of states graph for (A) pure silicene (B) CO2 on top of Siup (C) CO2 on top of Sidn.
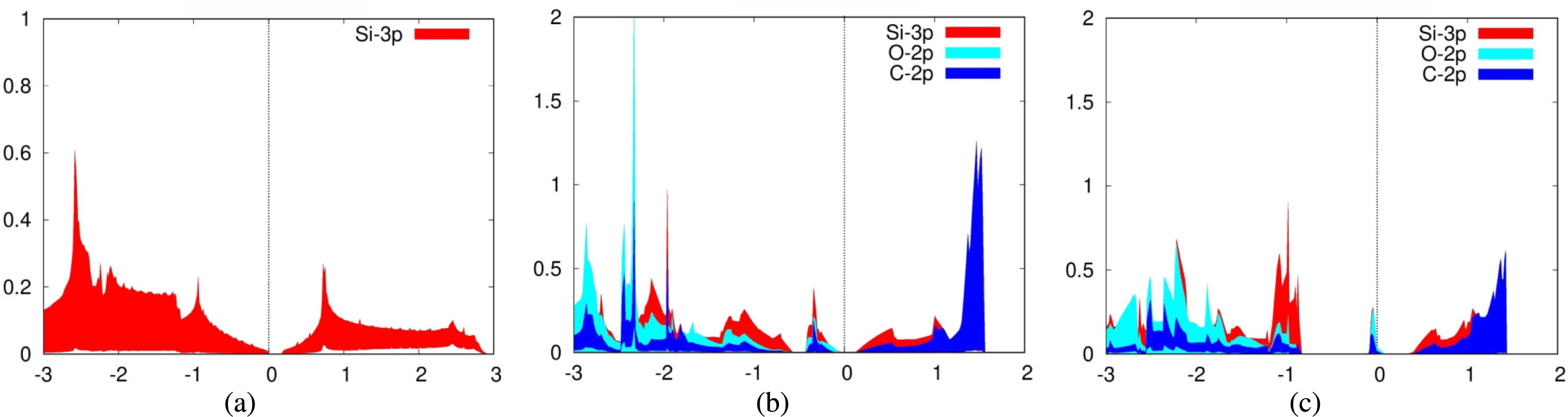{kind=link}

Figure 7: Charge transfer of carbon dioxide molecules on monolayer (teal atoms are silicon, yellow atoms are carbon, red atoms are oxygen; charge transfer plots: blue is charge lost, red is charge gained) the isosurface value we used was 0.3 e/Bohr3 (A) parallel to the Siup atoms of silicene (B) parallel to the Sidn atoms of silicene.
{kind=link}
Charge transfer
The formation of chemical bonds is further supported by the visible charge transfer in Fig. 7. When we plotted the charge transfer, we observed that both the Siup and Sidn atoms lost charge, while the oxygen atoms in carbon dioxide gained charge. Oxygen is more electronegative than silicon (1.9 for silicon vs. 3.44 for oxygen) because it only needs two more electrons to fill its valence shell, while silicon needs to gain or lose four electrons to achieve a full valence shell. The presence of charge transfer signifies that bonding did in fact occur between the carbon dioxide molecule and Siup and Sidn atoms.
Discussion
Throughout our calculations, we tested the adsorption of carbon dioxide on three different monolayers: pure aluminum nitride, defected aluminum nitride and pure silicene. Of the three, silicene has a buckled structure opposed to the planar structure of aluminum nitride. In addition, silicene also differs in atomic composition by being the only homoatomic monolayer contrasting with the heteroatomic compositions of aluminum nitride. We observed the effects of varying structure and composition on the adsorption capabilities of the monolayers for carbon dioxide specifically.
After processing our results, we noticed a few aspects of our research upon further analysis. First, we found that defected aluminum nitride had the most stable adsorption of carbon dioxide. However, carbon dioxide could be adsorbed onto all the different adsorption sites for silicene while the same was not true for aluminum nitride.
Carbon dioxide on pure aluminum nitride
Full relaxation was utilized for the adsorption of carbon dioxide onto aluminum nitride sheets. This allowed molecular free range of motion, meaning any bonds formed would be chemical bonds (Head & Zerner, 1985). We tested two different adsorption sites for carbon dioxide, although the molecule was only adsorbed over one site, when placed over the nitrogen atom. For the other site, over the aluminum atom, the carbon dioxide molecule did not show any binding affinity for the sheet as the molecule ran away. This led us to believe that carbon dioxide placed over the nitrogen atom was the only viable adsorption site for the aluminum nitride sheet. Further calculations (adsorption energy, PDOS, and charge transfer) were only calculated for this configuration of carbon dioxide on the aluminum nitride sheet.
After relaxation, we observed bonds between carbon dioxide and aluminum nitride, indicating a chemical adsorption of carbon dioxide on the aluminum nitride sheet. The configuration resulted in polarization of the carbon dioxide molecule, as charge was unevenly gained by the two oxygen atoms. Group-III nitride monolayers, such as aluminum nitride, tend to exhibit polarity because the nitrogen atom is more electronegative than the aluminum atom (Lebedev et al., 1999). This polarity could help adsorb the carbon dioxide molecule onto the sheet. The binding energies we observed were all negative, signifying a possible spontaneous adsorption.
The chemisorption of carbon dioxide onto aluminum nitride was further verified through both the PDOS and charge transfer. Strong hybridization between the interacting orbitals of the carbon dioxide molecule and aluminum nitride monolayer suggests that carbon dioxide was adsorbed onto aluminum nitride. Mainly, the 2p orbitals of nitrogen and the 2p orbitals of oxygen show this hybridization which makes sense because the oxygen atoms are being bonded to the nitrogen atoms.
Carbon dioxide on defected aluminum nitride
While carbon dioxide was not able to bond into all the adsorption sites of pure aluminum nitride, it was able to form bonds when placed on top of defected aluminum nitride. Creating a nitrogen vacancy resulted in polarization of the carbon dioxide molecule, as charge was unevenly gained by the two oxygen atoms. the success of the defected aluminum nitride sheet as opposed to the pure surface could be attributed to the fact that aluminum is an electron-deficient element and can only form three bonds. In the hexagonal arrangement of aluminum nitride, aluminum already possesses a full valence shell, making bonding over all adsorption sites very unlikely. When a nitrogen vacancy is performed, this frees one of aluminum’s bonds, allowing for the adsorption of carbon dioxide.
Carbon dioxide on pure silicene
Silicene was unique from the other two monolayers because it has a buckled structure, with distinct Siup and Sidn positions. This structural difference resulted in surface chemistries that contrasted with those of aluminum nitride. Full relaxation was utilized on this surface to determine the adsorption of carbon dioxide onto the sheet. The two oxygen atoms of carbon dioxide were found to have bonded to the Siup atoms. Even when we placed the carbon dioxide molecule over the Sidn atoms, the structure changed after relaxation and the Sidn atoms became Siup atoms. Previous studies have shown that within the dimer formed by the Siup and Sidn of a buckled silicene structure, charge was transferred from the Sidn atoms to the Siup atoms, leaving the Siup atoms with a lower energy (Li et al., 2013). Because of this charge transfer, the Siup atoms are associated with the highest occupied molecular orbit. The highest occupied molecular orbital is above the Siup atoms, characterized by an sp3 hybridization. With the high buckled silicene structure, the Sidn atoms tend to be more reactive, causing more electropositive elements to be adsorbed by the Siup atoms and more electronegative elementes tend to bind to the Sidn atoms.
However, our research utilized the use of low buckled silicene structure. The electronic properties of this structure differs slightly from those of the high buckled silicene structure (Luo et al., 2008). We inferred that the charge transfer between the Sidn and Siup atoms were not complete, leaving neither atom completely empty or completely full. This could result in the Sidn atoms being less reactive than those of a high buckled silicene structure. Furthermore, the Siup atoms could be more reactive than we initially hypothesized. This would allow electronegative species such as oxygen to bond to the Siup atoms. The adsorption of carbon dioxide onto silicene sheets was found to be negative, signifying a possible spontaneous adsorption. In addition, carbon dioxide was able to bind onto all the adsorption sites for silicene, unlike aluminum nitride. The difference could be attributed to the effects of silicene’s buckled structure on its surface chemistry. The results we observed are in agreement with previous studies as silicene is known to be an extremely reactive monolayer, and oftentimes it will spontaneously oxidize in the air (Vogt et al., 2012). Furthermore, S-O bonds are stronger than both Al-O and N-O bonds, with a bond energy of 809.0 kJ/mol, as compared to 501.9 kJ/mol and 631.6 kJ/mol for Al-O and N-O respectively (Brown & Shannon, 1973).
Conclusion
In summary, we tested three different 2D-materials, aluminum nitride, defected aluminum nitride, and silicene, for their ability to adsorb carbon dioxide. We performed first-principles calculations based on the density functional theory and utilized a full relaxation total energy technique. We analyzed adsorption energy, projected density of states, and charge transfer to determine the likelihood of adsorption occurring. The negative adsorption energy, strong hybridization of interacting atoms, and visible charge transfer led us to believe that carbon dioxide could be adsorbed on our monolayers. In addition, we observed the effects of different structures or atomic compositions on the surface chemistry of the monolayers. Our conclusions concluded that carbon dioxide can be adsorbed onto aluminum nitride over the nitrogen atom, defected aluminum nitride over both the aluminum and nitride atoms, and silicene over both the Siup and Sidn atoms.
In order to determine better adsorbent materials for carbon dioxide, future research could focus on doping and creating vacancies within the monolayers that we used. Since our study only preformed theoretical calculations to determine the adsorption for carbon dioxide on different 2D-materials, future studies could focus on the applications of this research using different 2D-materials to create devices to help adsorb carbon dioxide. In addition, our research looked only into the initial binding of carbon dioxide onto each of the monolayers. We did not consider whether or not the binding is reversible. Instead, we looked at how the binding of carbon dioxide changes the band structure of monolayers. Future research could focus on determining whether or not these binding configurations are permanent.