
Evolution of Wolbachia mutualism and reproductive parasitism: insight from two novel strains that co-infect cat fleas
- Published
- Accepted
- Received
- Academic Editor
- Ilaria Negri
- Subject Areas
- Entomology, Evolutionary Studies, Genomics, Microbiology, Zoology
- Keywords
- Wolbachia, Ctenocephalides felis, Cat flea, Reproductive parasitism, Mutualism, Lateral gene transfer, Cytoplasmic incompatibility, Biotin operon
- Copyright
- © 2020 Driscoll et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2020. Evolution of Wolbachia mutualism and reproductive parasitism: insight from two novel strains that co-infect cat fleas. PeerJ 8:e10646 https://doi.org/10.7717/peerj.10646
Abstract
Wolbachiae are obligate intracellular bacteria that infect arthropods and certain nematodes. Usually maternally inherited, they may provision nutrients to (mutualism) or alter sexual biology of (reproductive parasitism) their invertebrate hosts. We report the assembly of closed genomes for two novel wolbachiae, wCfeT and wCfeJ, found co-infecting cat fleas (Ctenocephalides felis) of the Elward Laboratory colony (Soquel, CA, USA). wCfeT is basal to nearly all described Wolbachia supergroups, while wCfeJ is related to supergroups C, D and F. Both genomes contain laterally transferred genes that inform on the evolution of Wolbachia host associations. wCfeT carries the Biotin synthesis Operon of Obligate intracellular Microbes (BOOM); our analyses reveal five independent acquisitions of BOOM across the Wolbachia tree, indicating parallel evolution towards mutualism. Alternately, wCfeJ harbors a toxin-antidote operon analogous to the wPip cinAB operon recently characterized as an inducer of cytoplasmic incompatibility (CI) in flies. wCfeJ cinB and three adjacent genes are collectively similar to large modular toxins encoded in CI-like operons of certain Wolbachia strains and Rickettsia species, signifying that CI toxins streamline by fission of large modular toxins. Remarkably, the C. felis genome itself contains two CI-like antidote genes, divergent from wCfeJ cinA, revealing episodic reproductive parasitism in cat fleas and evidencing mobility of CI loci independent of WO-phage. Additional screening revealed predominant co-infection (wCfeT/wCfeJ) amongst C. felis colonies, though fleas in wild populations mostly harbor wCfeT alone. Collectively, genomes of wCfeT, wCfeJ, and their cat flea host supply instances of lateral gene transfers that could drive transitions between parasitism and mutualism.
Introduction
Wolbachiae (Alphaproteobacteria: Rickettsiales: Anaplasmataceae) comprise Gram-negative, obligate intracellular bacteria that infect over half the world’s described insect species as well as certain parasitic nematodes (Hilgenboecker et al., 2008). Unlike other notable rickettsial genera that contain human pathogens (e.g., Rickettsia, Orientia, Neorickettsia, Anaplasma, and Ehrlichia), wolbachiae do not infect vertebrates (Gillespie et al., 2012). A single species, Wolbachia pipientis, is formally recognized with numerous members designated as strains within 16 reported supergroups (Augustinos et al., 2011; Baldo & Werren, 2007; Bing et al., 2014; Glowska et al., 2015; Haegeman et al., 2009; Lo et al., 2002; Ros et al., 2009; Lefoulon et al., 2020). Genomic divergence indicates further species names are warranted (Chung et al., 2018), though increasing diversity and community consensus suggest caution regarding further Wolbachia classification at the species level (Lindsey et al., 2016; Ramírez-Puebla et al., 2015).
Like other obligate intracellular microbes, wolbachiae are metabolic parasites that complement a generally reduced metabolism with pilfering of host metabolites (Driscoll et al., 2017; Jiménez et al., 2019). Their ability to survive and flourish is also heavily influenced by the acquisition of key functions through lateral gene transfer (LGT). Several described Wolbachia strains demonstrate characteristics of limited mutualism with their invertebrate partner (Zug & Hammerstein, 2015; Hosokawa et al., 2010), through the synthesis and provisioning of riboflavin (Moriyama et al., 2015) and biotin (Ju et al., 2019; Nikoh et al., 2014). While riboflavin biosynthesis genes are highly conserved in wolbachiae (Newton & Rice, 2020), biotin biosynthesis genes are rare and likely originated via LGT with taxonomically divergent intracellular bacteria (Gillespie et al., 2012). Still other strains of Wolbachia exert varying degrees of reproductive parasitism (RP) on their insect host (Werren, Baldo & Clark, 2008), influencing host sexual reproduction via processes such as male-killing, feminization, parthenogenesis and cytoplasmic incompatibility (CI) (Hurst & Frost, 2015). Wolbachia genes underpinning CI and male killing have been characterized (Chen et al., 2019; Beckmann et al., 2019; LePage et al., 2017; Beckmann, Ronau & Hochstrasser, 2017; Beckmann & Fallon, 2013; Perlmutter et al., 2019) and occur predominantly in the eukaryotic association module (EAM) of Wolbachia prophage genomes (Bordenstein & Bordenstein, 2016). These genes highlight the role of LGT in providing wolbachiae with factors facilitating mutualism or RP, both of which are highly successful strategies for increasing infection frequency in invertebrate host populations.
Compared to reproductive parasites, Wolbachia mutualists appear to form more stable, long-term relationships with their hosts, as supported by Wolbachia-host codivergence in certain filarial nematodes (Bandi et al., 1998) and Nomada bees (Gerth & Bleidorn, 2017). In contrast to the stability of mutualists, relationships of reproductive parasites appear more ephemeral. RP can be a strong mechanism to increase infection frequency; for example, hosts of CI-inducing strains can rapidly replace uninfected members within populations (Turelli & Hoffmann, 1991; Schmidt et al., 2017). Despite this, CI is prone to neutralization through the evolution of host suppression (Hurst & Frost, 2015; Cooper et al., 2017; Turelli, 1994) and purifying selection on the host does not preserve CI (Turelli, 1994).
Recent work proposes that CI genes, or CI factors (cifs), have genetic life cycles (Martinez et al., 2020). Initially, cifs might colonize new hosts by LGT, pushing said host to high frequency in populations through CI. Ultimately, CI collapses as cifs become pseudogenized. Thus, cifs are selfish elements that persist in a manner similar to transposons (Martinez et al., 2020; Cooper et al., 2019). The occurrence of duplicate cif copies (i.e., encoding the same biochemical mechanism), as well as cif pseudogenes, in many Wolbachia strains supports this model (Gillespie et al., 2018). Cif degradation would lead to a weak CI background, fomenting favorable conditions for Wolbachia strains with more active cif repertoires to invade and outpace resident strains carrying failing cifs. An alternative to autonomous cif jumping would be recombinant WO-phages or smaller elements actually carrying CI operons. If dosage is important to CI, all these events might shift the strength of CI leading to new bidirectional incompatibilities.
Such LGT partly explains why Wolbachia estimated phylogenies are often discordant with those of their hosts (Cooper et al., 2019; Raychoudhury et al., 2009; Turelli et al., 2018). Horizontal transmissions of Wolbachia strains, which can occur through direct host interactions, environmental contacts (shared habitat, food sources, etc.) or predator/parasitoid delivery, also cause tree discordance (Pietri, DeBruhl & Sullivan, 2016) and contribute to episodic invasions, replacements, and exchange of cifs. Horizontal transmission is evident in cases where hosts are infected with multiple divergent Wolbachia strains (Raychoudhury et al., 2009; Turelli et al., 2018; Pietri, DeBruhl & Sullivan, 2016). In host populations that vary from single- vs double-infections, co-infection might reflect a transitory phase (i.e., one strain replacing another) or stable co-infection. We envision that competition could arise between co-infections if the distinct reproductive parasites carry too similar RP-inducing genes and battle for control of parallel reproductive mechanisms. Alternatively, perhaps the stability of co-infections could be supported in part by different mechanistic approaches to RP (e.g., cin vs cid loci) or dosage effects from multiple strains collaborating to instantiate a new crossing type. Furthermore, stable coinfections might increase robustness by partitioning distinct strains to serve within distinct host micro-niches (i.e., nutrient provisioning vs RP); each might increase fitness distinctly or alternatively parasitize distinctly. Such a case is in part witnessed in doubly-infected cochineals (Dactylopius coccus), wherein a Supergroup B strain (wDacB) carries an arsenal of RP-inducing genes but a Supergroup A strain (wDacA) carries none (Raychoudhury et al., 2009). Clearly, documenting more cases of Wolbachia co-infections putatively serving under contrasting host relationships will be helpful to understand mechanisms of replacement and stability in host populations.
We recently sequenced the genome of the cat flea (Ctenocephalides felis) and concurrently closed the genomes of two novel wolbachiae, wCfeT and wCfeJ, that co-infect fleas of the Elward Laboratory (Soquel, CA, USA; hereafter EL fleas) (Driscoll et al., 2020). Genome-based phylogeny estimation placed wCfeT on a basal divergent branch, while wCfeJ was found to subtend Wolbachia Supergroup C. Both genomes show evidence of past WO phage infection, with LGT hotspots indicating recent acquisition of factors predicted to either underpin mutualism (wCfeT) or mediate CI (wCfeJ). Additionally, we describe two loci within the C. felis genome itself that support repeated acquisitions of CI antidote genes by the host. Infection surveys of cat flea populations and colonies from across the US indicate extensive spread of wCfeT among wild populations, and a strong bias for wCfeT/wCfeJ co-infection in colonies, highlighting the complex ecological relationship that drives this system. Finally, we apply an informatics approach including phylogenomics, synteny analysis, and functional alignment to construct a comprehensive picture revealing the recurrent evolution of nutritional symbiosis and RP across the Rickettsiales.
Materials and Methods
Assembly and annotation of wCfeT and wCfeJ
Our recent report detailed (1) the assembly of Wolbachia reads generated from C. felis genome sequencing, (2) genome circularization and naming of two novel strains (wCfeT and wCfeJ), and (3) complete genome annotation (Driscoll et al., 2020). All relevant information pertaining to wCfeT and wCfeJ is available at NCBI under Bioproject PRJNA622233. Complete genome sequences are available in the NCBI RefSeq database at accession identifiers NZ_CP051156.1 (wCfeT) and NZ_CP051157.1 (wCfeJ).
Wolbachiae phylogenomics
Protein sequences (n = 84,836) for 66 sequenced Wolbachia genomes plus five additional Anaplasmataceae (Neorickettsia helminthoeca str. Oregon, Anaplasma centrale Israel, A. marginale Florida, Ehrlichia chaffeensis Arkansas, and E. ruminantium Gardel) were either downloaded directly from NCBI (n = 48), retrieved as genome sequences from the NCBI Assembly database (n = 13), contributed via personal communication (n = 8; Michael Gerth, Oxford Brookes University), or sequenced here (n = 2) (Table S1). For genomes lacking functional annotations (n = 15), gene models were predicted using the RAST v2.0 server (n = 12) or GeneMarkS-2 v1.10_1.07 (n = 3; (Lomsadze et al., 2018)). Ortholog groups (n = 3,038) were subsequently constructed using FastOrtho, an in-house version of OrthoMCL (Li, Stoeckert & Roos, 2003), using previously established criteria (i.e., an expect threshold of 0.01, percent identity threshold of 30%, and percent match length threshold of 50% for ortholog inclusion (Driscoll et al., 2017, 2020; Smith et al., 2015; Hagen et al., 2018)). A subset of single-copy families (n = 12) conserved across at least 60 of the 66 genomes were independently aligned with MUSCLE v3.8.31 (Edgar, 2004) using default parameters. Regions of poor alignment were masked with trimal v1.4.rev15 (Capella-Gutiérrez, Silla-Martínez & Gabaldón, 2009) using the “automated1” option. All modified alignments were concatenated into a single data set (2,803 positions) for phylogeny estimation using RAxML v8.2.4 (Stamatakis, 2014). The gamma model of rate heterogeneity and estimation of the proportion of invariant sites were used. Branch support was assessed with 1,000 pseudo-replications. Final ML optimization likelihood was −52350.085098.
BOOM characterization
Phylogeny estimation
Genomes (n = 93) containing at least four of the six biotin synthesis genes were identified using Blastp searches against the NCBI nr database (e-value < 0.01; accessed 9 January 2020). Protein sequences for these loci were downloaded from NCBI, aligned with MUSCLE v3.8.31 (Edgar, 2004) using default parameters, and regions of poor alignment masked with trimal v1.4.rev15 (Capella-Gutiérrez, Silla-Martínez & Gabaldón, 2009) using the “automated1” option. All modified alignments were concatenated into a single data set (1,457 positions) for phylogeny estimation using RAxML v8.2.4 (Stamatakis, 2014), under the gamma model of rate heterogeneity and estimation of the proportion of invariant sites. Branch support was assessed with 1,000 pseudo-replications. Final ML optimization likelihood was −127140.955066. See Table S2 for NCBI protein accession IDs for all sequences included in this analysis.
Gene neighborhood analysis of wCfeT BOOM
Genes in a 25-gene window around the wCfeT BOOM cluster were used to query (Blastp) the NCBI nr database (accessed 9 January 2020). The top 50 matches to each sequence (e-value < 1, BLOSUM45 matrix) were binned by taxonomic assignment as “Wolbachia”, “other Rickettsiales”, or “other (non-Rickettsiales) taxa.” Other (non-Rickettsiales) taxa were further binned by major taxonomic group to assess the boundaries and diversity of the wCfeT BOOM MGE.
Synteny analysis of biotin genes in wolbachiae
Rickettsiales genomes with at least 5 of the 6 biotin synthesis genes (n = 28) were downloaded from NCBI and ortholog groups (n = 2,527) constructed with FastOrtho, an in-house version of OrthoMCL (Li, Stoeckert & Roos, 2003), using an expect threshold of 0.01, percent identity threshold of 30%, and percent match length threshold of 50% for ortholog inclusion. OGs containing biotin synthesis genes were identified using Rickettsia buchneri str. Wikel sequences as seeds. Genome locations for all genes were retrieved from NCBI in gene file format (gff) and used to order the orthologs in each genome.
Phylogeny estimation of the EamA protein of wCfeT
Taxa were selected from Blastp searches against the NCBI Rickettsiales and nr (excluding Rickettsiales) databases using the wCfeT EamA protein (WP_168464633.1) as a query. Proteins were aligned with MUSCLE v3.8.31 (Edgar, 2004) using default parameters, with regions of poor alignment masked using Gblocks (Talavera & Castresana, 2007). Phylogeny was estimated with the WAG substitution model using RAxML v8.2.4 (Stamatakis, 2014) under the gamma model of rate heterogeneity and estimation of the proportion of invariant sites.
CI gene characterization
Wolbachiae
wCfeJ CinA and CinB were compared to other RP-inducing TA operons following our previous approach (Gillespie et al., 2018). Another novel CndAB operon was identified on a small scaffold (NCBI Nucleotide ID CABPRJ010001055.1) in the aphid Cinara cedri, which has an associated Wolbachia symbiont, wCed (Gómez-Valero et al., 2004), and was also characterized in a similar manner. Briefly, EMBL’s Simple Modular Architecture Research Tool (SMART) (Letunic & Bork, 2017) and /or the Protein Homology/analogY Recognition Engine V 2.0 (Phyre2) (Kelley & Sternberg, 2009) were used to predict the following domains: OTU cysteine protease (Pfam OTU, PF02338) (Makarova, Aravind & Koonin, 2000), endoMS-like (Nakae et al., 2016) and NucS-like (Ren et al., 2009) PD-(D/E)XK nuclease (Pfam PDDEXK_1, PF12705) (Kinch et al., 2005), CE clan protease (Pfam Peptidase_C1, PF00112) (Pruneda et al., 2016), Latrotoxin-CTD (Zhang et al., 2012), and ankyrin repeats. Individual protein schemas were generated using Illustrator of Biological Sequences (Liu et al., 2015) with manual adjustment.
C. felis
A cidA-like gene was identified in the Ctenocephalides felis genome during initial genome decontamination (Driscoll et al., 2020). Briefly, a contig composed of a mosaic of flea- and Wolbachia-like sequence was identified by our comparative BLAST-based pipeline and flagged for manual inspection. Gene models on this contig were predicted using the RAST v2.0 server. The resulting proteins were used to create a custom Blast database that was queried using Blastp with CinAB sequences from Wolbachia endosymbiont of Culex quinquefasciatus Pel (CAQ54390, CAQ54391), and CidAB sequences Wolbachia pipientis wAlbB (CCE77512, CCE77513). The contig was subsequently confirmed as belonging to the largest scaffold (NW_020538040) in the C. felis assembly. A second, cinA-like gene was identified by querying all CDS in the published C. felis assembly (NCBI accession ASM342690v1) with the same set of Wolbachia CinAB and CidAB sequences.
Screening colonies and wild populations of cat fleas for wCfeT and wCfeJ
Upon receipt from collaborators, cat fleas were stored at −20 °C in EtOH until processing for DNA extraction and qPCR. Fleas were sexed under a stereomicroscope and surface sterilized for 5 min with 1% bleach solution, followed by 5 min with 70% EtOH, and finally with three washes using molecular grade water. Individual fleas were flash-frozen in liquid N2 and ground to powder with sterile pestle in a 1.5 ml microcentrifuge tube. DNA was extracted from dissected tissues using the Zymo Quick-DNA Miniprep plus kit following the solid tissue protocol (Zymo Research; Irvine, CA, USA). The presence of wCfeT, wCfeJ, and C. felis DNA was determined via qPCR using the following primer sets: Cfe#18S|179, 5′-TGCTCACCGTTTGACTTGG-3′ and 5′-GTTTCTCAGGCTCCCTCTCC-3′ (Reif et al., 2008); wCfeT#ApaG|75: 5′-GCCGTCACTGGCAGGTAATA-3′ and 5′-GCTGTTCTCCAATAACGCCA-3′, wCfeJ#CinA|76, 5′-AGCAACACCAACATGCGATT-3′ and 5′-GAACCCCAGAGTTGGAAGGG-3′. Each primer set amplicon was sequenced before use to ensure specificity. qPCR was performed using a QuantStudio 3 with the PowerUp SYBR green mastermix in a 20 μl reaction volume containing 2 μl of sample DNA with 400 nM of each primer. Cycling parameters were consistent with the “fast cycling mode” including a 2 min cycle at 50 °C, a 2 min cycle at 95 °C, 40 cycles with 3 s at 95 °C and 30 s at 60 °C followed by a melt curve analysis. Standard curves were generated to quantify the number of DNA copies present in each reaction. Results were expressed as the ratio of Wolbachia amplicons to C. felis 18S rDNA copies to normalize for varying efficiency of DNA extractions. Fleas were considered positive for wolbachiae if the flea 18S rDNA Ct value is 30 or lower and the Wolbachia Ct value is 37 or lower.
Determining wCfeT and wCfeJ localization in C. felis tissues
Elward Laboratories (EL, Soquel, CA, USA) cat fleas stored at −80 °C were used to assay tissue localization of wCfeT and wCfeJ. Ovaries and midguts of individual female fleas were dissected under a stereomicroscope. DNA extraction and qPCR were performed as described above.
Results and Discussion
Fleas are understudied vectors of several human and animal diseases (Rust, 2017). While wolbachiae (mostly unnamed strains) have been detected in many flea species (Gorham, Fang & Durden, 2003; Dittmar & Whiting, 2004; Lawrence et al., 2015), it is pressing from a biocontrol perspective to know if these bacteria affect the ability of fleas to vector pathogens. The cat flea transmits pathogenic bacteria such as Rickettsia typhi (murine typhus), R. felis (murine typhus-like illness), Bartonella spp. (e.g., cat-scratch disease and various other bartonelloses), and to a lesser extent the plague agent Yersinia pestis (Glickman et al., 2006; Nogueras et al., 2013; Angelakis et al., 2016; Leulmi et al., 2014). Cat fleas are also intermediate hosts of the tapeworm Dipylidium caninum and the filarial nematode Acanthocheilonema reconditum, both of which can infect humans (Fourie et al., 2012; Brianti et al., 2012; Traversa, 2013). Other organisms observed in C. felis include undescribed Baculoviridae, amebae, trypanosomatids, cephaline gregarines (Apicomplexa), and microsporidia (Beard, Butler & Hall, 1990). However, nearly all epidemiological studies have focused on determining the frequency and distribution of only certain pathogens (e.g., Bartonella spp., R. typhi, R. felis), with few documenting wolbachiae co-occurrence with these pathogens (Tay, 2013; Oteo et al., 2014).
Identification of novel Wolbachia parasites
Recently, we assembled and annotated closed genomes from two divergent Wolbachia strains, named wCfeT and wCfeJ, that were captured during genome sequencing of the cat flea (EL Colony) (Driscoll et al., 2020). The 16S rDNA sequences of these two strains are identical to those previously identified in another cat flea colony (Louisiana State University) (Pornwiroon et al., 2007; Sunyakumthorn et al., 2008; Gillespie et al., 2015), which historically has been replenished with EL colony fleas. wCfeT and wCfeJ are broadly similar to other closed Wolbachia genomes in terms of length, GC content, number of protein coding genes, and coding density (Table 1), although it is interesting to note that wCfeT exhibits one of the highest pseudogene counts (second only to wCle) while wCfeJ boasts one of the lowest (second only to wOv). Coupled with other recently sequenced genomes for non-Supergroup A and B strains (e.g., wFol and wVulC), these cat flea-associated wolbachiae blur the lines demarcating genomic traits previously used to distinguish basal and sister Wolbachia lineages from those of Supergroups A and B strains (Lefoulon et al., 2020), particularly regarding genome size and mobile element composition (Lefoulon et al., 2020).
| Taxon | RefSeq ID | SG* | Length (nt) | GC (%) | Protein coding genes | Pseudogenes | Mean gene length (nt) | Coding density (coding/total nt) |
|---|---|---|---|---|---|---|---|---|
| wFol | NZ_CP015510.2 | E | 1801626 | 34.4 | 1537 | 85 | 976 | 0.880 |
| wCfeT | NZ_CP051156.1 | ? | 1495538 | 35.2 | 1424 | 201 | 890 | 0.727 |
| wOv | NZ_HG810405.1 | C | 960618 | 32.1 | 650 | 45 | 967 | 0.700 |
| wOo | NC_018267.1 | C | 957990 | 32.1 | 639 | 73 | 968 | 0.719 |
| wCfeJ | NZ_CP051157.1 | ? | 1201647 | 35.6 | 1116 | 51 | 917 | 0.813 |
| wCle | NZ_AP013028.1 | F | 1250060 | 36.3 | 1012 | 210 | 818 | 0.829 |
| wBm | NZ_CP034333.1 | D | 1080064 | 34.2 | 803 | 178 | 868 | 0.789 |
| wBm | NC_006833.1 | D | 1080084 | 34.2 | 839 | 167 | 854 | 0.795 |
| wMau | NZ_CP034334.1 | B | 1273527 | 34.0 | 1045 | 136 | 944 | 0.876 |
| wMau | NZ_CP034335.1 | B | 1273530 | 34.0 | 1047 | 131 | 946 | 0.876 |
| wNo | NC_021084.1 | B | 1301823 | 34.0 | 1066 | 133 | 952 | 0.878 |
| wAlbB FL2016 | NZ_CP041923.1 | B | 1482279 | 34.5 | 1190 | 195 | 917 | 0.858 |
| wAlbB HN2016 | NZ_CP041924.1 | B | 1483853 | 34.4 | 1200 | 187 | 917 | 0.858 |
| wAlbB | NZ_CP031221.1 | B | 1484007 | 34.4 | 1197 | 191 | 916 | 0.858 |
| wMeg | NZ_CP021120.1 | B | 1376868 | 34.0 | 1136 | 125 | 948 | 0.869 |
| wHa | NC_021089.1 | A | 1295804 | 35.1 | 1123 | 104 | 899 | 0.852 |
| wCauA | NZ_CP041215.1 | A | 1449344 | 35.0 | 1260 | 134 | 861 | 0.860 |
| wAna | NZ_CP042904.1 | A | 1401460 | 35.2 | 1218 | 107 | 893 | 0.857 |
| wRi | NC_012416.1 | A | 1445873 | 35.2 | 1245 | 122 | 892 | 0.859 |
| wAu | NZ_LK055284.1 | A | 1268461 | 35.2 | 1099 | 125 | 878 | 0.855 |
| wMel | NC_002978.6 | A | 1267782 | 35.2 | 1116 | 129 | 863 | 0.857 |
| wMel I23 | NZ_CP042444.1 | A | 1269137 | 35.2 | 1119 | 128 | 862 | 0.857 |
| wMel N25 | NZ_CP042446.1 | A | 1267781 | 35.2 | 1120 | 125 | 863 | 0.857 |
| wMel ZH26 | NZ_CP042445.1 | A | 1267436 | 35.2 | 1117 | 125 | 865 | 0.857 |
| wIrr | NZ_CP037426.1 | A | 1352354 | 35.3 | 1232 | 115 | 827 | 0.855 |
Note:
Prior reports have indicated the presence of multiple Wolbachia “types” infecting C. felis, with phylogenies inferred from partial gene sequences placing these unnamed Wolbachia strains in supergroups B or F, or in basal lineages (Gorham, Fang & Durden, 2003; Dittmar & Whiting, 2004; Casiraghi et al., 2005). Our robust genome-based phylogeny estimation reveals that both wCfeT and wCfeJ are divergent from Supergroup A and B wolbachiae (Fig. 1; Table S1). wCfeT is similar to undescribed C. felis-associated strains that branch basally to most other Wolbachia lineages (Gorham, Fang & Durden, 2003; González-Álvarez et al., 2017; Vasconcelos et al., 2018), while wCfeJ is similar to undescribed C. felis-associated strains closely related to Wolbachia supergroups C, D and F (Gorham, Fang & Durden, 2003; Casiraghi et al., 2005). The substantial divergence of wCfeT and wCfeJ from a Wolbachia supergroup B strain associated with C. felis from Germany (wCte) indicates a diversity of wolbachiae that are capable of infecting cat fleas. Importantly, the nature of host-microbe relationships for any of these C. felis-associated wolbachiae is unknown.

Figure 1: Wolbachia genome-based phylogeny estimation.
Wolbachia supergroups are within colored ellipses. Ctenocephalides felis-associated Wolbachiae are within black boxes, with red (wCfeT) and blue (wCfeJ) stars depicting the two novel Wolbachia parasites infecting C. felis. Gray inset: color scheme for nematode and arthropod hosts. Phylogeny was estimated for 66 Wolbachia and five outgroup genomes (see “Materials and Methods” for more details). Branch support was assessed with 1,000 pseudoreplications. Final ML optimization likelihood was −52350.085098.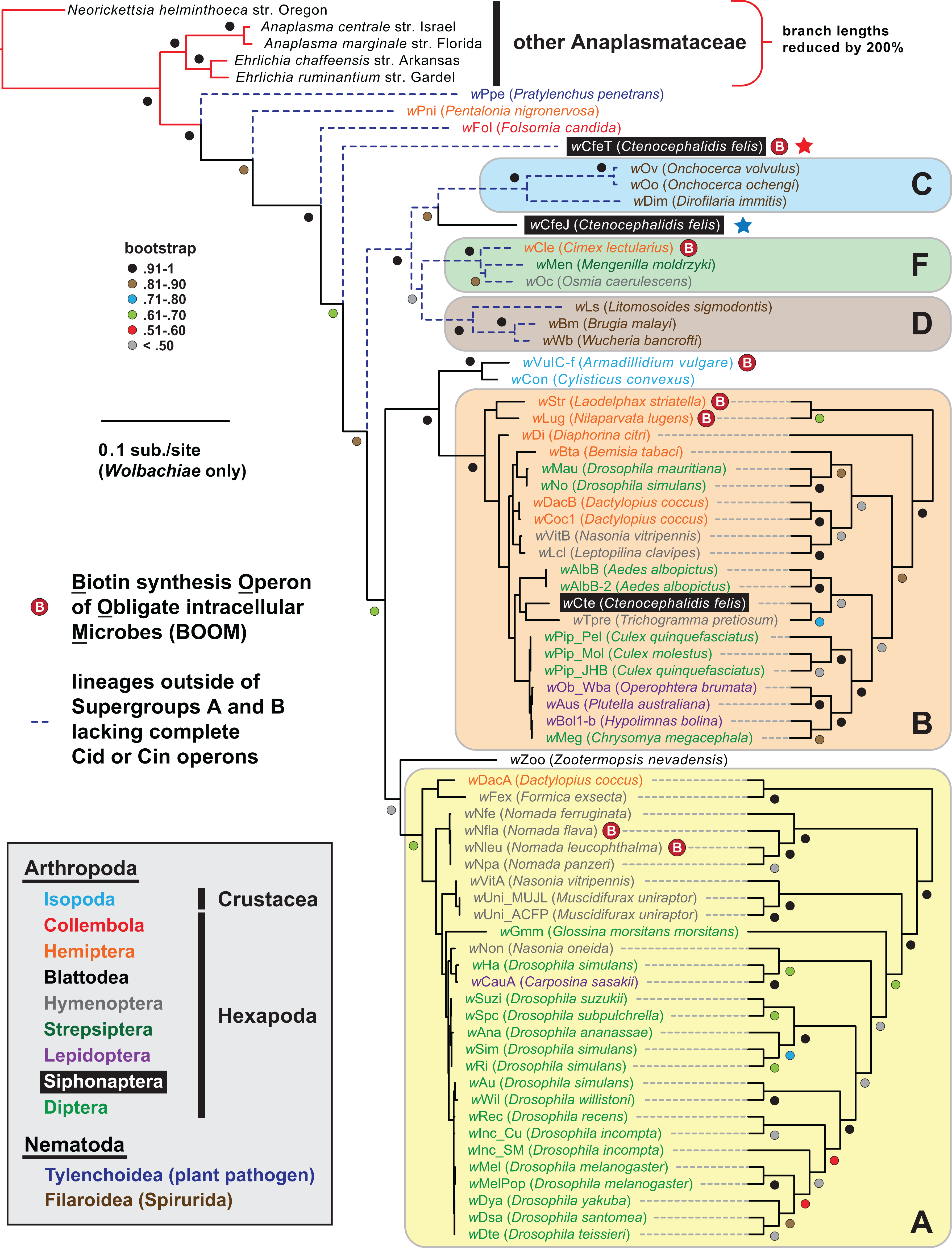{kind=link}
wCfeT is equipped with biotin synthesis genes
The enzymatic steps for bacterial biosynthesis of biotin from malonyl-CoA are largely conserved (Lin & Cronan, 2011) and usually involve six bio enzymes and the fatty acid biosynthesis machinery (Lin, Hanson & Cronan, 2010) (Fig. 2A). We previously identified the first plasmid-borne bio gene operon, which occurs on plasmid pREIS2 of Rickettsia buchneri, an endosymbiont of the black-legged tick (Gillespie et al., 2012). We observed a rare gene order for the pREIS2 bio operon shared only with bio operons encoded on the chromosomes of obligate intracellular pathogens Neorickettsia spp. and Lawsonia intracellularis (Deltaproteobacteria). These unique bio operons formed a clade in estimated phylogenies, indicating LGT of the operon across divergent intracellular species. Subsequent studies, using our same dataset for phylogeny estimation, discovered this bio operon in certain Wolbachia (Nikoh et al., 2014; Gerth & Bleidorn, 2017; Balvín et al., 2018), Cardinium (Penz et al., 2012; Zeng et al., 2018) and Legionella (Ríhová et al., 2017) species. We refer hereafter to this intriguing LGT of bio genes as the Biotin synthesis Operon of Obligate intracellular Microbes (BOOM).
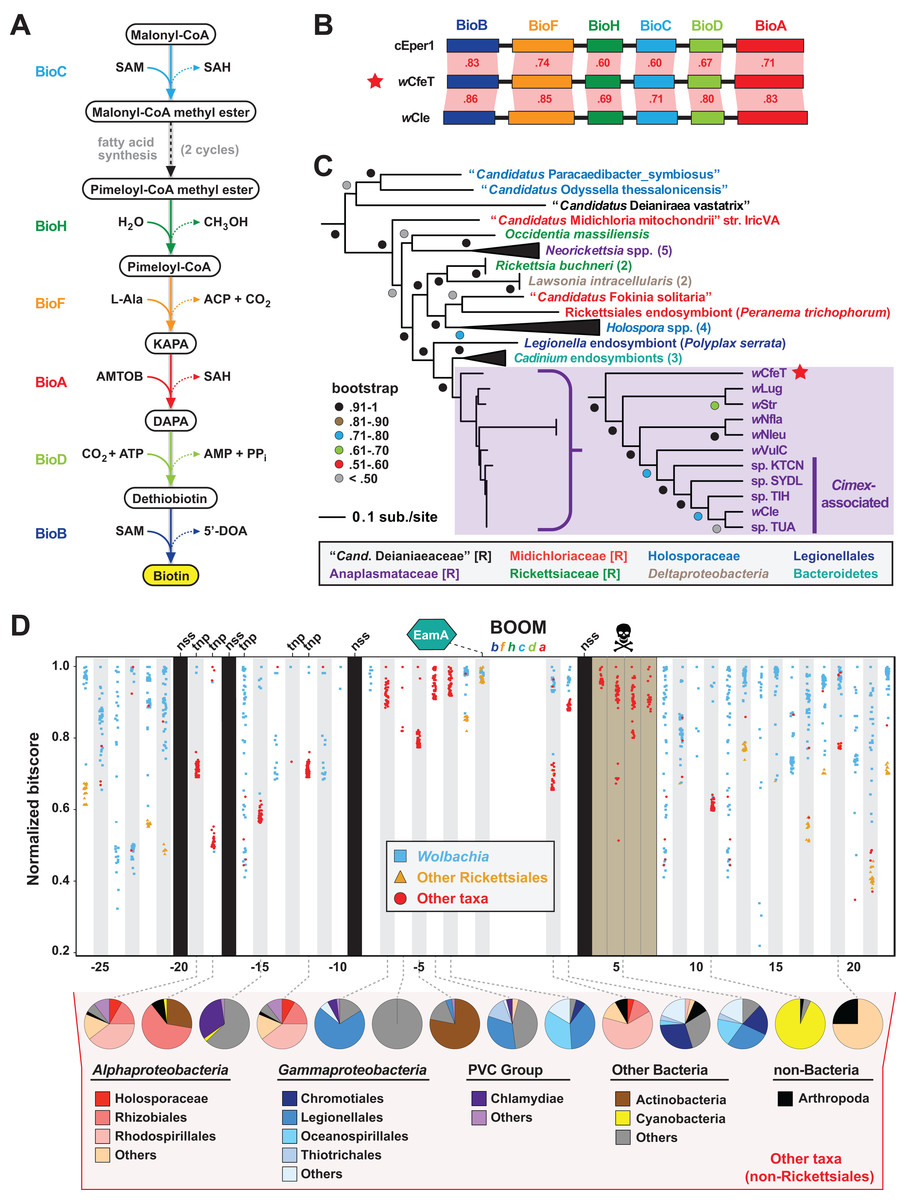
Figure 2: wCfeT carries the Biotin synthesis Operon of Obligate intracellular Microbes (BOOM).
(A) Metabolism of biotin from malonyl-CoA (Lin & Cronan, 2011). (B) Comparisons of wCfeT BOOM to equivalent loci in Cardinium endosymbiont of Encarsia pergandiella (cEper1, CCM10336–CCM10341) and Wolbachia endosymbiont of Cimex lectularius (wCle, BAP00143–BAP00148). Red shading and numbers indicate % identity across pairwise protein alignments (Blastp). Gene colors correspond to enzymatic steps depicted in panel A, with all proteins drawn to scale (as a reference, wCfeT BioB is 316 aa). (C) Phylogeny of BOOM and other bio gene sets from diverse bacteria estimated from the concatenation of six bio enzymes (BioC, BioH, BioF, BioA, BioD, and BioB); see Table S2 for all sequence information, and Fig. S1A for complete tree estimated under Maximum Likelihood. In taxon color scheme, [R] denotes Rickettsiales, with Holosporaceae as a revised family of Rhodospirillales (Muñoz-Gómez et al., 2019). (D) Analysis of genes flanking wCfeT BOOM. See Table S3 for comparison with other BOOM-containing wolbachiae. Graph depicts top 50 Blastp subjects (e-value < 1, BLOSUM45 matrix) using the wCfeT proteins as queries against the NCBI nr database (January 9, 2020). Colors distinguish three taxonomic groups (see inset). Black columns: no significant similarity (nss) in the NCBI nr database. Tnp, predicted transposase. Brown, pseudogenization. See Fig. S1B for EamA phylogeny estimation. Pie charts across the bottom delineate taxonomic diversity of hits to “Other taxa,” for wCfeT proteins with significant Blastp matches outside of the Rickettsiales.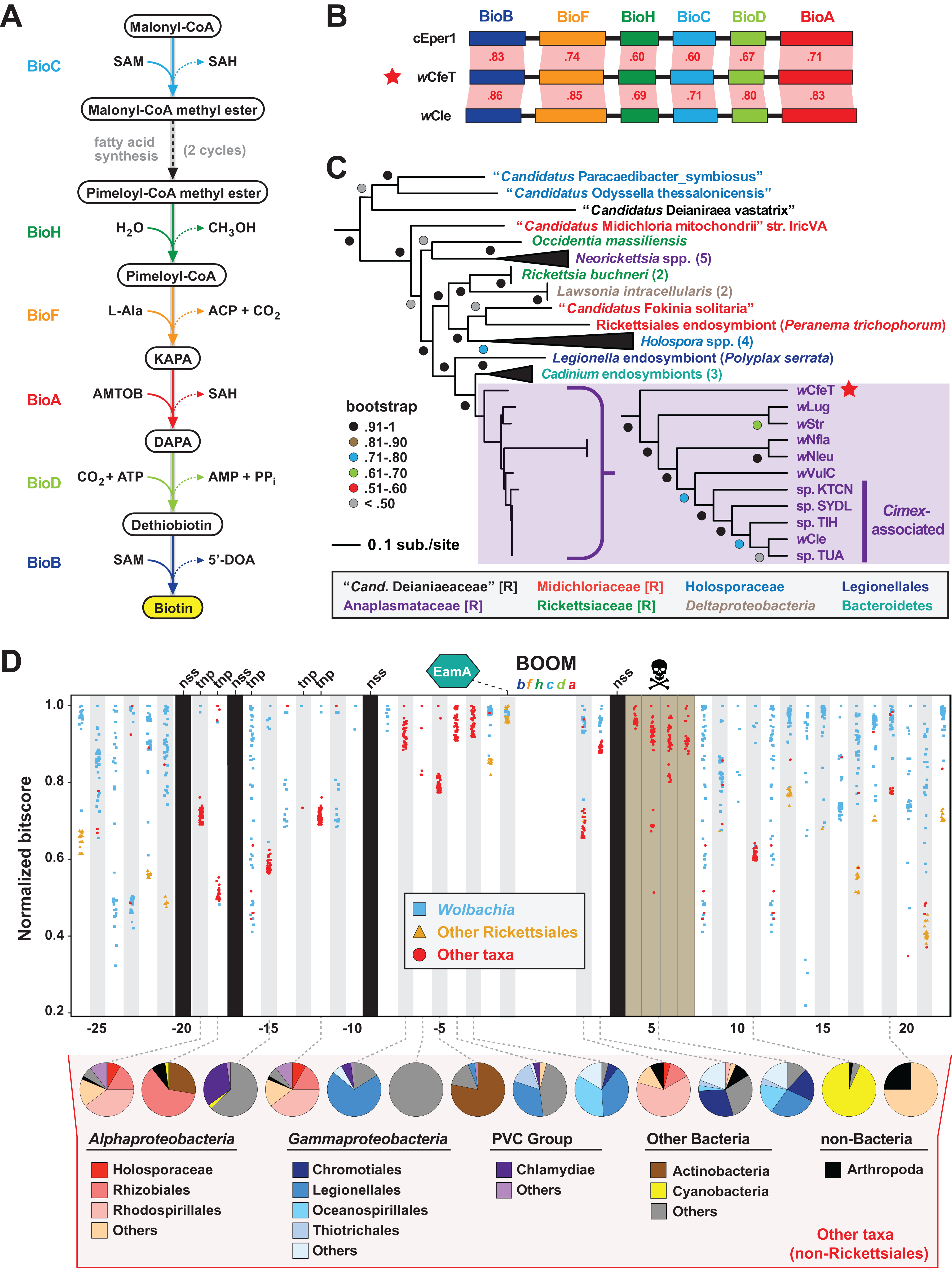{kind=link}
wCfeT carries a complete BOOM with greater similarity to Wolbachia BOOM than those of other bacteria (Fig. 2B). Phylogeny estimation of BOOM and other diverse bio gene sets indicates wCfeT BOOM is basal to other Wolbachia BOOM (Fig. 2C; Fig. S1). However, as the divergence pattern for other Wolbachia BOOM is discordant with genome-based phylogeny (Fig. 1), it is clear that multiple independent BOOM acquisitions have occurred in wolbachiae irrespective of lineage divergences. This is supported by a previous study positing recent BOOM acquisition in wNfla and wNleu (Gerth & Bleidorn, 2017) and a current report that detected partial BOOM loci in wApol, a Supergroup S Wolbachia strain infecting pseudoscorpions (Lefoulon et al., 2020). The nature of BOOM flanking regions in the genomes of wCfeT, wVulC, wCle, wStr, and wLug, which collectively lack synteny and are riddled with transposases, recombinases and phage-related elements (Table S3), also attests to LGT as the origin for BOOM in these genomes. Furthermore, analysis of wCfeT BOOM flanking genes reveals hotspots for additional LGT (Fig. 2D), exemplified by an S-adenosylmethionine importer (EamA) gene (Fig. S1B) that is a signature among many diverse obligate intracellular bacteria (Gillespie et al., 2012; Klinges et al., 2019; Haferkamp et al., 2013; Driscoll et al., 2013).
As some BOOM-containing Wolbachia strains have been shown to provide biotin to their insect hosts (Ju et al., 2019; Nikoh et al., 2014), we posit that wCfeT has established an obligate mutualism with C. felis mediated by biotin-provisioning. Genes involved in CI (and putatively in MK) have been predominantly found in the EAM; the absence of such genes in wCfeT’s WO prophage (Fig. S2) provides no evidence that wCfeT can induce these two forms of RP, implying that its maintenance may indeed rely on nutritional supplementation to the cat flea. Wolbachia strains associated with bedbug (wCle), planthopper (wStr, wLug), and termites (wCtub) with evidence for BOOM (Hellemans et al., 2019) reside in bacteriocytes of their insect hosts, structures associated with nutritional symbiosis in many arthropods (Douglas, 2015). Interestingly, C. felis is not known to contain bacteriocytes, and the ability of wCfeT to synthesize and supplement biotin to cat fleas awaits experimentation.
wCfeJ lends insight on the evolution of reproductive parasitism
wCfeJ carries a putative toxin-antidote (TA) operon that is architecturally similar to the wPip_Pel CinA/B TA operon (Fig. 3A), which was previously characterized as a CI inducer in flies (Chen et al., 2019). Containing at least three distinct variants (LePage et al., 2017; Lindsey et al., 2018), all CinB toxins harbor dual nuclease (NUC) domains in place of the deubiquitinase (DUB) domain of CidB, another CI inducer in flies (LePage et al., 2017; Beckmann, Ronau & Hochstrasser, 2017; Beckmann & Fallon, 2013). Chimeric NUC-DUB toxins, termed CndB toxins (Beckmann et al., 2019), also occur as operons (cndAB) in certain Wolbachia and Rickettsia genomes (Beckmann, Ronau & Hochstrasser, 2017; Gillespie et al., 2018). Although their functions remain unknown, these larger CndB toxins belong to an extraordinarily diverse array of toxins with highly modular architectures that occur in divergent intracellular bacteria (Gillespie et al., 2018). Curiously, despite a size similar to the CinB toxins, wCfeJ’s CinB toxin has much higher sequence identity to larger CndB and NUC-containing toxins found mostly in Wolbachia Supergroups A and B (Fig. 3B). A similar pattern was found for wCfeJ’s CinA antidote, indicating the genes were likely acquired as an operon.
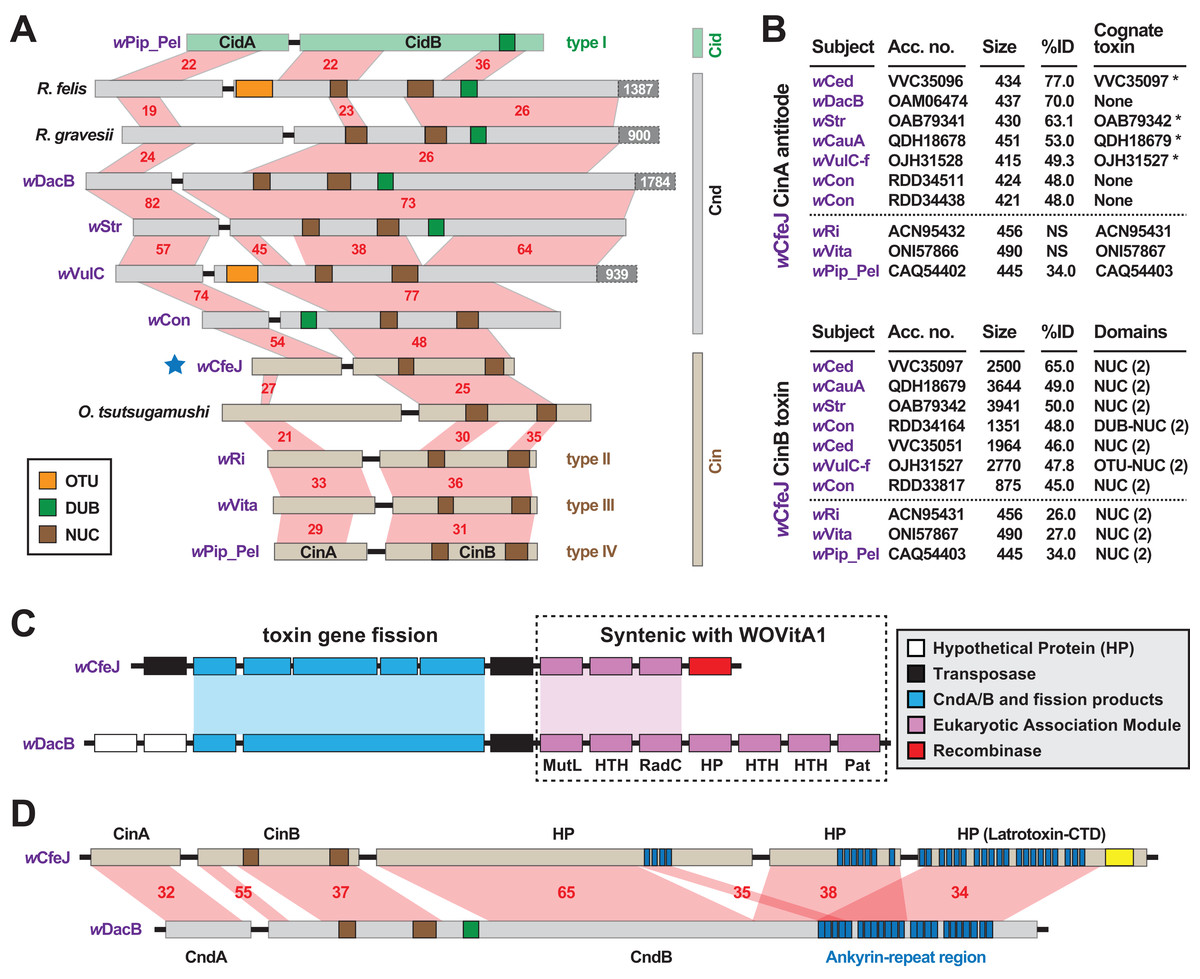
Figure 3: wCfeJ lends insight on the evolution of reproductive parasitism.
(A) Comparison of known and predicted RP-inducing toxin-antidote operons of diverse rickettsial species. Light green: CidA/B of wPip_Pel (CAQ54390, CAQ54391); gray: CndA/B of pLbAR_36/38 (KHO02168, KHO02170), Rickettsia gravesii (WP_017442886, WP_024547315), wDacB (OAM06111, OAM06112), wStr (OAB79364, OAB79365), wVulC (OJH31528, OJH31527), wCon (RDD34163, RDD34164); light brown, CinA/B of wCfeJ (blue star, WP_168456002 & WP_168456001), Orientia tsutsugamushi (CAM80637, CAM80639), wRi (ACN95432, ACN95431), wVita (ONI57866, ONI57867), and wPip_Pel (CAQ54402, CAQ54403). Type I-IV CI operons of Wolbachia parasites (LePage et al., 2017) are distinguished. Protein domains as follows: green, CE clan protease; brown, PD-(D/E)XK nuclease; orange, OTU cysteine protease. Red shading and numbers indicate % identity across pairwise alignments. Proteins are drawn to scale, with additional C-terminal sequence for some CndB toxins depicted in dark gray boxes. (B) Sequence similarity profiles for wCfeJ’s CinA antidote (top) and CinA toxin (bottom). Subjects are from a Blastp search against the NCBI “Wolbachia” database. Size, amino acids. Antidotes with cognate toxins (operon partners) also recovered in searches are noted with asterisks. Top seven hits are shown, with matches (where significant) to proteins of types II-IV TA operons shown below dashed lines. Toxin domains are from our prior report or predicted newly here using the EMBL’s Simple Modular Architecture Research Tool (SMART) (Letunic & Bork, 2017). Only NUC and DUB domains are listed, though larger toxins contain many other predicted domains. (C) Comparison of wCfeJ and wDacB degraded WO prophage genomes captures streamlining of a CI-like TA operon from a Cnd operon. WO prophage coordinates: wCfeJ, WP_168455994–WP_168456003; wDacB, OAM06111–OAM06118 (LSYY01000098). Schema follows the description of the WOVitA prophage (Bordenstein & Bordenstein, 2016). (D) wCfeJ’s CinB-like toxin evolved from gene fission of a larger CndB-like toxin. NCBI protein accession numbers: wCfeJ (WP_168455999, WP_168456000, and WP_168456001); wDacB (OAM06111, OAM06112). Red shading and numbers indicate % identity across pairwise alignments. Proteins are drawn to scale; as a reference, wDacB CndB is 3707 aa.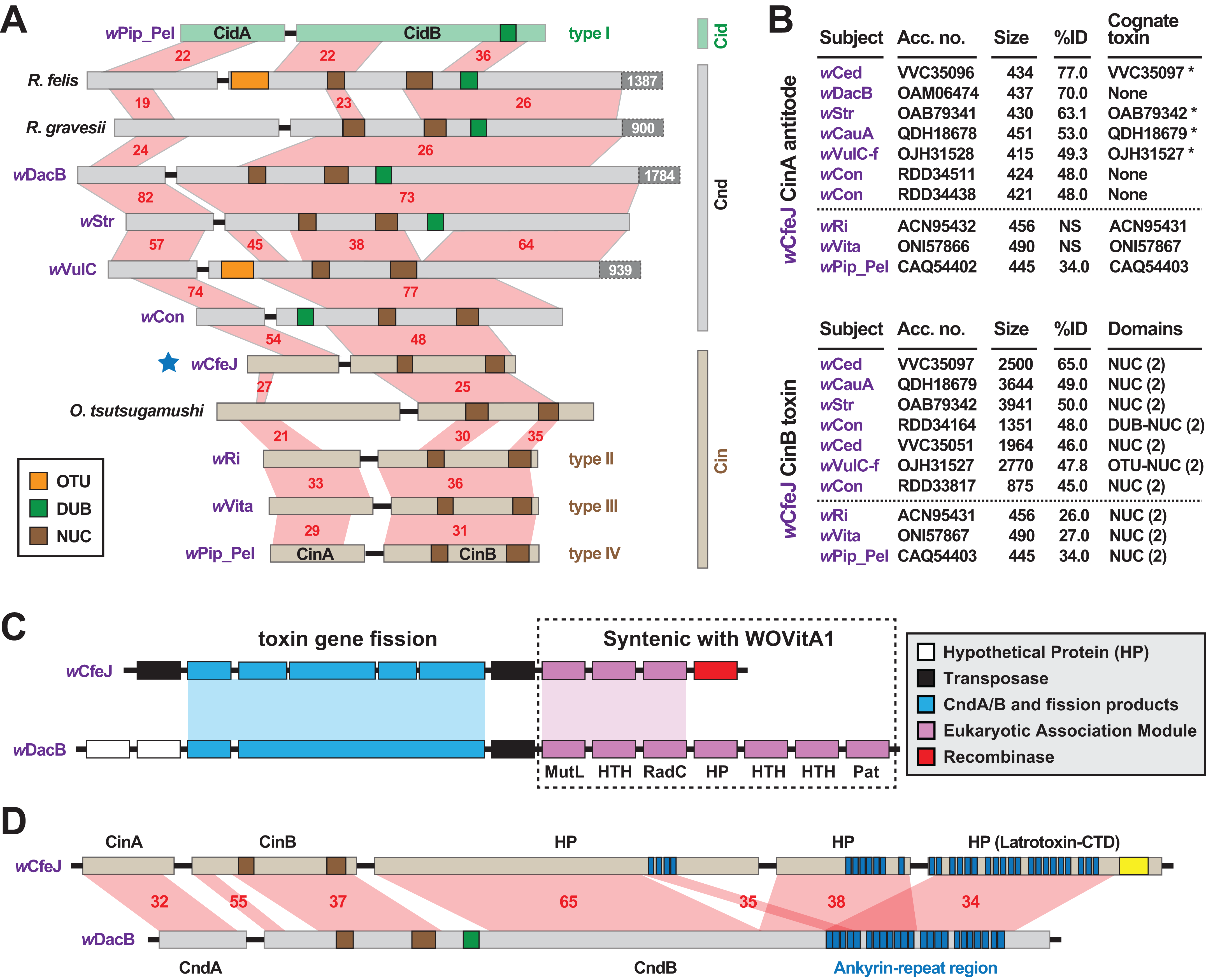{kind=link}
In contrast to wCfeT, wCfeJ harbors a highly degraded WO prophage genome with only a partial EAM, the CinA/B operon, and several other genes encoding hypothetical proteins and transposases (Fig. 3C). This is reminiscent of the CI- and male killer-inducing wRec strain, which lacks a functional WO phage yet possesses TA modules within remnant EAMs (Metcalf et al., 2014).
Synteny analysis with other Wolbachia phages revealed wCfeJ cinB and three downstream genes are colinear with a large (3707 aa) wDacB cndB toxin (Fig. 3D). We posit that gene fission of a large modular CndB toxin created the smaller CinB toxin. This interpretation is consistent with our prior hypothesis stating that more streamlined RP-inducing toxins (i.e., cinB and cidB) originate from larger modular toxins (i.e., cndB and others) that are widespread in the intracellular mobilome, particularly Wolbachia phage and Rickettsia plasmids and integrative and conjugative elements (ICEs) (Gillespie et al., 2018, 2015). We find the alternative hypothesis, that multiple gene fusions create larger modular toxins like CndB proteins, less parsimonious provided that RP toxins tend to degrade and pseudogenize rapidly in rickettsial genomes (Martinez et al., 2020; Gillespie et al., 2018).
wCfeJ is unique among described wolbachiae outside of Supergroups A and B by containing a CI-like TA operon. Some of the Wolbachia strains in closely related Supergroups C, F and D are considered mutualists with their nematode or arthropod hosts, supporting the absence of RP-inducing genes in the genomes of these strains. Still, other strains not included in our analysis, namely those belonging to Supergroups S (Lefoulon et al., 2020; Chafee et al., 2010), P (Glowska et al., 2015), and J (Lefoulon et al., 2016) may eventually be shown to carry RP-inducing genes, especially since some of these strains are not known as mutualists. wFol, another basal Wolbachia lineage (Fig. 1), does carry a myriad of large modular candidate RP-inducing toxins but does not have cin, cid or cnd TA operons (Faddeeva-Vakhrusheva et al., 2017; Kampfraath et al., 2019). This indicates LGT of candidate RP-inducing genes occurs across the breadth of Wolbachia diversity. Collectively, we speculate that wCfeJ acquired a WO prophage, possibly from a Supergroup A or B Wolbachia strain that also infects cat fleas (e.g., wCte), and that viral- or gene-level selection streamlined a cndAB locus into a cinAB operon (Gillespie et al., 2018). Given that the genomes of many Wolbachia reproductive parasites harbor diverse arrays of CinA/B-and CidA/B-like operons (Gillespie et al., 2018; Beckmann et al., 2019), we posit that wCfeJ’s CinA/B TA operon might function in CI or some other form of RP; however, further study is required to evaluate wCfeJ as a cat flea reproductive parasite. Furthermore, an intriguing conjecture is that functions of the larger modular toxins, exemplified by wDacB CndB, whatever they be, are likely the primal functions that gave rise to CI.
Lateral transfer of Wolbachia CI-like antidotes to the cat flea genome
Our previous work identified CI-like toxin and antidote genes on scaffolds in several arthropod genome assemblies, although antidote genes were found more frequently possibly due to utility in suppressing microbial invaders implementing RP (Gillespie et al., 2018). Many of these genomes contain multiple divergent antidote genes, though none were found to harbor large-scale Wolbachia gene transfer that is known to occur in certain invertebrates (Leclercq et al., 2016; Hotopp et al., 2007; Aikawa et al., 2009; Kondo et al., 2002; Dhaygude et al., 2019; McNulty et al., 2010; Werren et al., 2010). In light of this observation, we originally speculated that CI-like genes are mobilized from wolbachiae to host through WO phage insertion into host nuclear genomes; however, the absence of other WO phage genes in these genomes made this assertion tenuous. Instead, more recent work describing the first WO prophage carrying two sets of CI loci provides compelling evidence for transposon-mediated LGT of CI loci between divergent wolbachiae (Cooper et al., 2019). This phage-independent mechanism for RP gene jumping in wolbachiae may result in the inadvertent capture of these genes by arthropod host genomes.
When we analyzed the cat flea genome for CI-like genes, we discovered that the largest C. felis scaffold (185.5 Mb) was found to harbor two CI-like antidote genes (Fig. 4A). Remarkably, neither antidote has any significant similarity to CinA of wCfeJ (Fig. 4B), indicating that these CI antidotes originated from other, as yet undescribed wolbachiae. The C. felis CinA-like CDS (XP_026474240.1) has greater similarity to CinA proteins than CidA proteins, while the CidA-like CDS (not annotated in the current C. felis assembly) has much greater similarity to CidA antidotes (Beckmann, Ronau & Hochstrasser, 2017; Beckmann & Fallon, 2013; Bonneau et al., 2018; Madhav et al., 2020; Morrow et al., 2020). The C. felis cinA-like gene is found as an exon within a larger flea gene model (LOC113377978), and is well supported (18× coverage, 7.2 TPM) by transcripts from the 1KITE project (Misof et al., 2014). Since these transcripts were generated in fleas from a different colony (Dr. Michael Dryden, Kansas State University), we conclude that this gene transfer occurred during an historical infection predating the establishment of these two colonies. Finally, the C. felis CinA- and CidA-like antidotes each possess the conserved motifs we previously defined for all CI antidotes (Gillespie et al., 2018), with the exception of a truncation of motifs 5 and 6 in the CinA-like protein (Fig. 4C). As other arthropod-encoded antidotes share this C-terminal truncation, it may signify an expendable domain in the host (i.e., a bacterial secretion signal, a motif involved in cognate toxin activation or delivery, etc.).
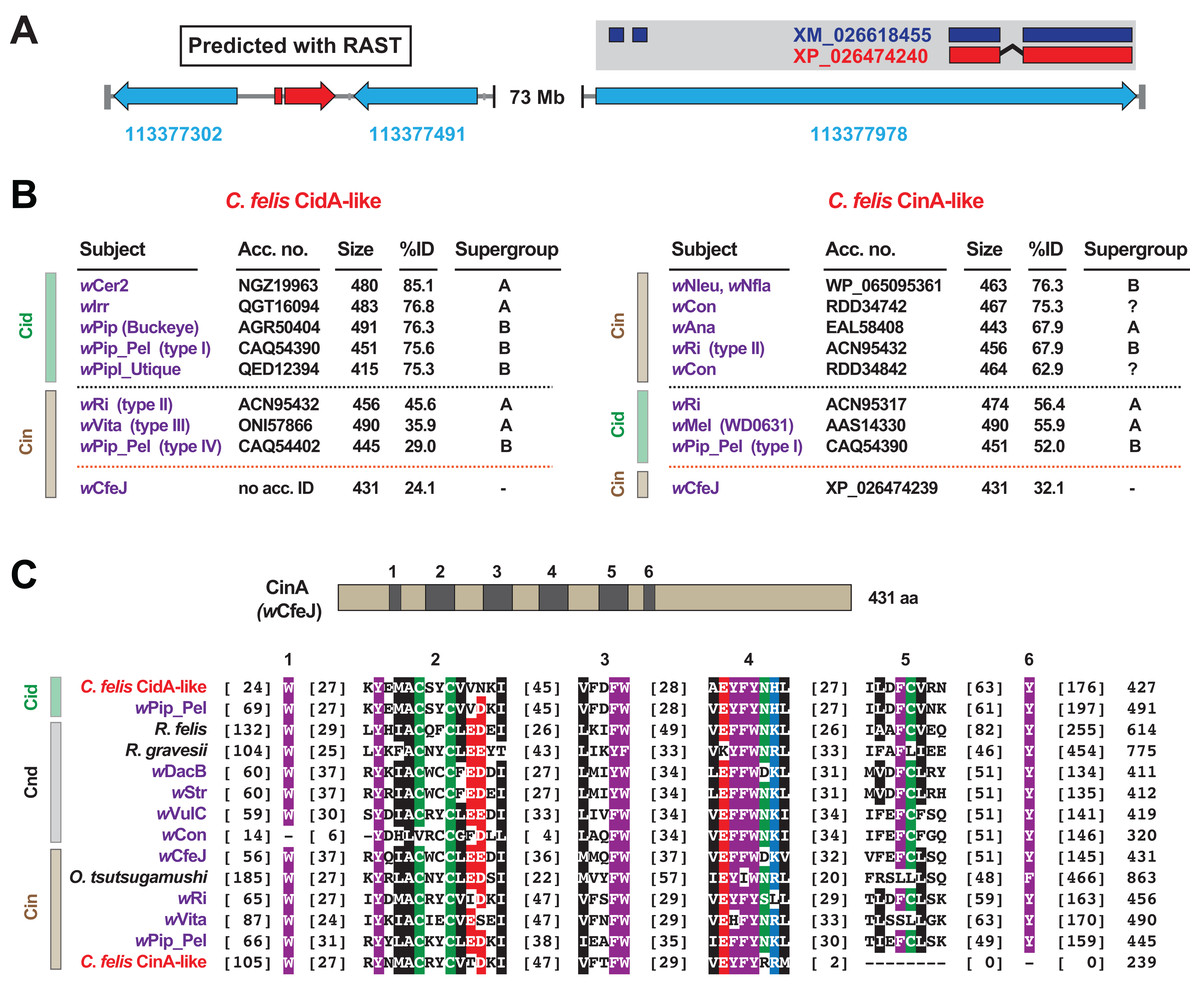
Figure 4: Identification of two CI-like antidote genes in the cat flea genome.
(A) The largest C. felis scaffold (NW_020538040) contains a CidA-like gene, predicted using RAST (Aziz et al., 2008) (left), and a CinA-like exon encoded within a larger gene model with transcriptional support (right). Red, CI-like antidote genes. Blue, C. felis genes: 113377302 (RING finger and SPRY domain-containing protein 1-like); 113377491 (negative elongation factor B); 113377978 (uncharacterized). The CinA-like exon (XP_026474240) contains a corresponding transcript (dark blue, XM_026618455) generated from a study (Misof et al., 2014) independent of the cat flea genome sequencing (Driscoll et al., 2020). (B) Sequence similarity profiles for the C. felis CidA-like (left) and CinA-like (right) antidotes. Subjects above the black dashed line are the top hits from a Blastp search against the NCBI nr protein database; subjects below are weaker matches to CinA (left) and CidA (right) proteins; matches to wCfeJ CinA are below the orange line. Size, amino acids. (C) Alignment of C. felis CidA-like and CinA-like antidotes to CidA, CndA and CinA proteins from selected Rickettsiaceae species and Wolbachia strains. Schema at top depicts the wCfeJ CinA antidote and location of six conserved regions shown in the alignment below. Amino acid coloring as follows: black, hydrophobic; red, negatively charged; green, hydrophilic; purple, aromatic; blue, positively charged. Alignment generated using MUSCLE, default parameters (Edgar, 2004).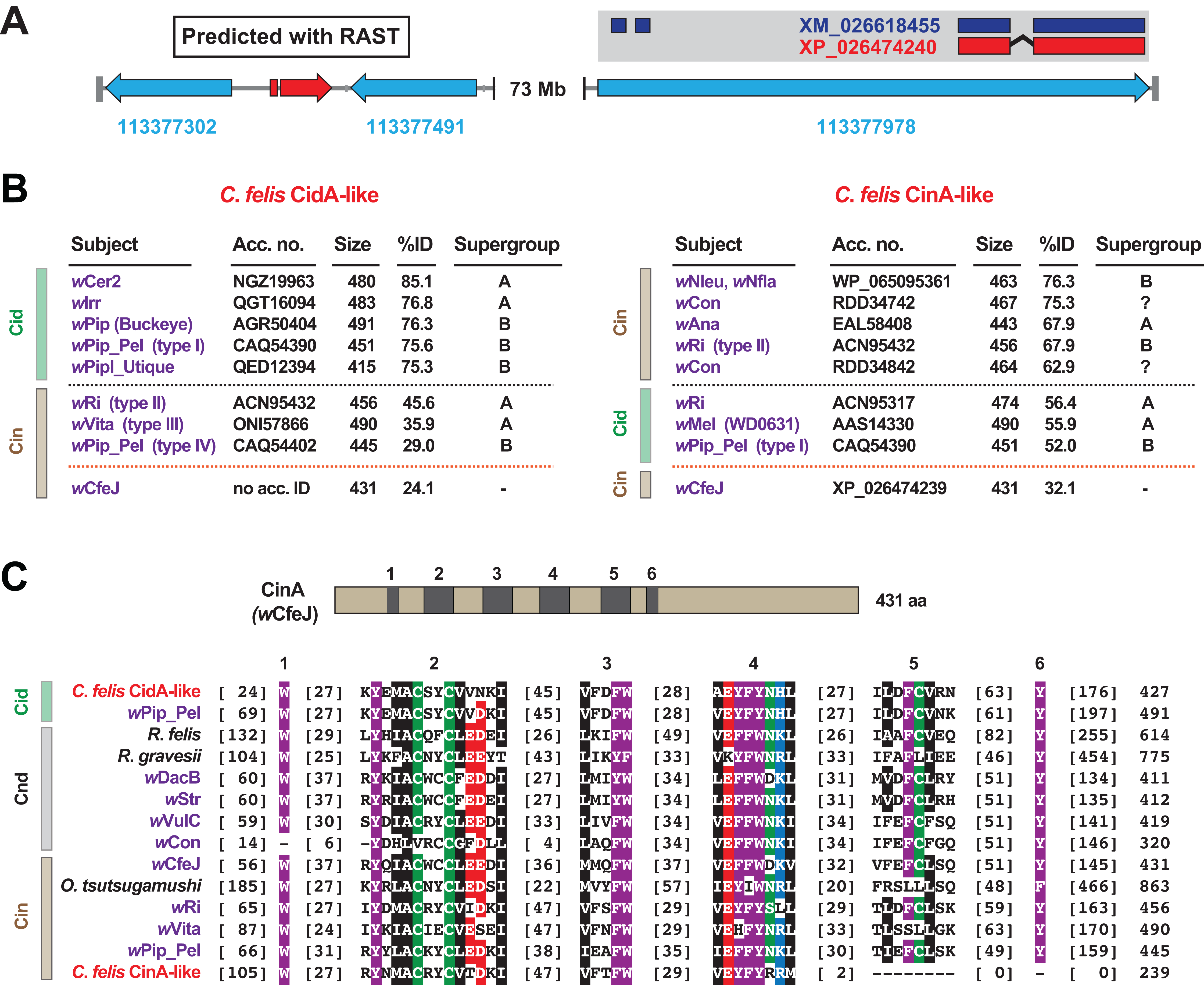{kind=link}
The incorporation of a Wolbachia antidote into an exon of a host gene provides one explanation for how such a laterally transferred gene would remain functional in the absence of the native regulatory elements encoded on WO prophage genomes. We previously hypothesized that CI antidote genes would be under strong selection in arthropod genomes if their expression could result in rescuing CI induction by reproductive parasites (Gillespie et al., 2018). In support of this supposition, it has been shown that: host genotypes modulate CI (Metcalf et al., 2014; Hornett et al., 2006, 2014; Bordenstein, Uy & Werren, 2003); some hosts show weak (Dmel and D. suzukii (Cooper et al., 2017; Conner et al., 2017; Hamm et al., 2014)) or strong (Culex pipiens and D. simulans (Poinsot, Charlat & Merçot, 2003; Merçot & Charlat, 2004)) CI; and a Wolbachia strain’s CI can be suppressed when transfected across different hosts (Bordenstein, Uy & Werren, 2003). While none of the arthropod-encoded CI antidotes have been characterized, we propose they impart immunity to toxins secreted by intracellular parasites, curtailing chronic parasite drive into populations. At least four independent evolutionary mechanisms might lead to suppression of CI. Gene family expansion of, and/or overexpression of key factors capable of counteracting CI mechanisms is possible; similarly, it was postulated that evolution in Cid interacting proteins like karyopherin-α or P32 could lead to target site resistance (Beckmann et al., 2019). A third possibility contributing to weakening CI, though not suppression per se, is simply the mutational collapse of the operons themselves due to lack of purifying selection (Martinez et al., 2020; Meany et al., 2019). We supply evidence of a fourth possibility whereby CI might be counteracted by germline expression of acquired CI antidotes. Significantly, our finding raises awareness of a potential barrier to Wolbachia-mediated pathogen biocontrol measures.
Characterization of wCfeT and wCfeJ infection of cat fleas
Coupled with earlier reports indicating multiple early-branching wolbachiae lineages infecting C. felis (Gorham, Fang & Durden, 2003; Casiraghi et al., 2005), our characterization of two divergent Wolbachia strains infecting cat fleas in the EL colony (Driscoll et al., 2020) as well as fleas maintained for a decade at LSU (Pornwiroon et al., 2007; Sunyakumthorn et al., 2008; Gillespie et al., 2015) prompted us to test individual fleas for double infection. In addition to EL fleas, three other colony strains and three geographically diverse wild cat flea populations were also surveyed (Fig. 5A). Aside from the Modesto Wild strain, which is historically replenished with wild-caught cat fleas (Dr. B. Donahue, 2017, personal communication), strong wCfeJ-wCfeT co-infection was found in colony fleas. Conversely, single wCfeT infection dominated wild cat fleas, with only a few fleas from a large sampling throughout Orange Co. CA harboring wCfeJ. While far greater sampling of both colony and wild populations is necessary, our results hint at a strong selection for wCfeT over wCfeJ in nature, which is concordant with our premise that wCfeT’s BOOM might drive mutualism. The ability of wCfeJ to highly infect colony fleas may arise as a consequence of bottlenecks within colonies, which are known to strengthen CI and maternal artificial transmission (Turelli, 1994). No sex ratio distortions were observed in colony or wild fleas, ruling out other forms of RP as driving factors. Nonetheless, the ability of wCfeT and wCfeJ to co-infect individual cat fleas provides a system to study contrasting forces (nutritional symbiosis and RP) that potentially drive multiple Wolbachia infections in a single invertebrate host.
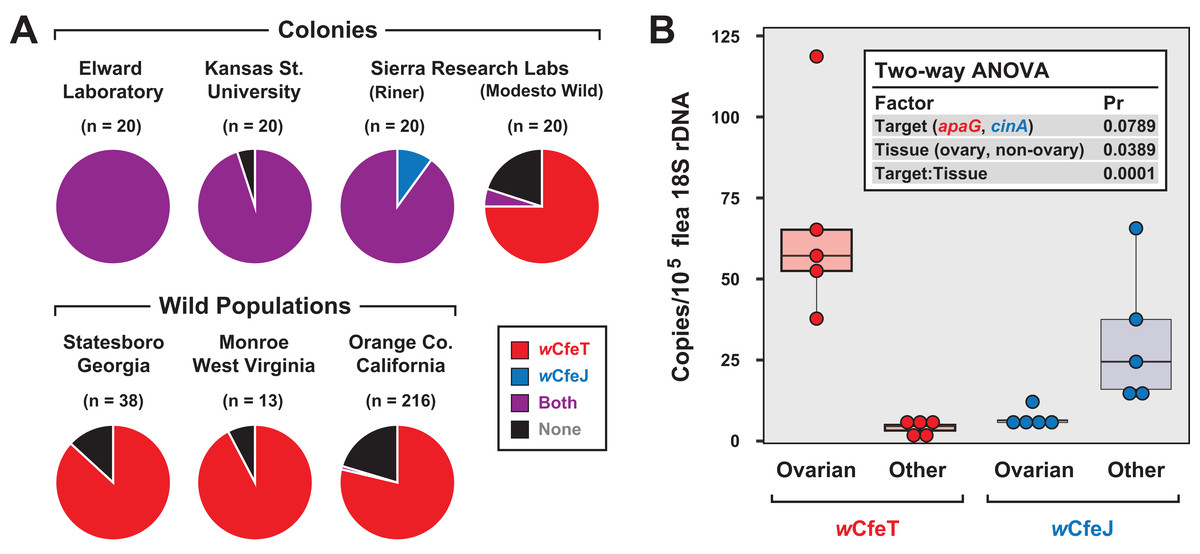
Figure 5: Characterization of wCfeJ and wCfeJ in cat fleas.
(A) Screening for wolbachiae in geographically diverse cat flea colonies (top) and wild populations (bottom). Cat fleas from six locations (including two strains from Sierra Research Laboratories) were assessed for the presence of wCfeT and wCfeJ. Wolbachia infection of individual fleas was assessed by qPCR (see “Materials and Methods” for details). (B) Localization of wCfeT and wCfeJ in cat flea tissues. Dissected ovaries and other tissues (mostly midguts) from individual cat fleas (EL Colony) were assayed by qPCR (see “Materials and Methods” for details). Five fleas were assayed for each tissue type.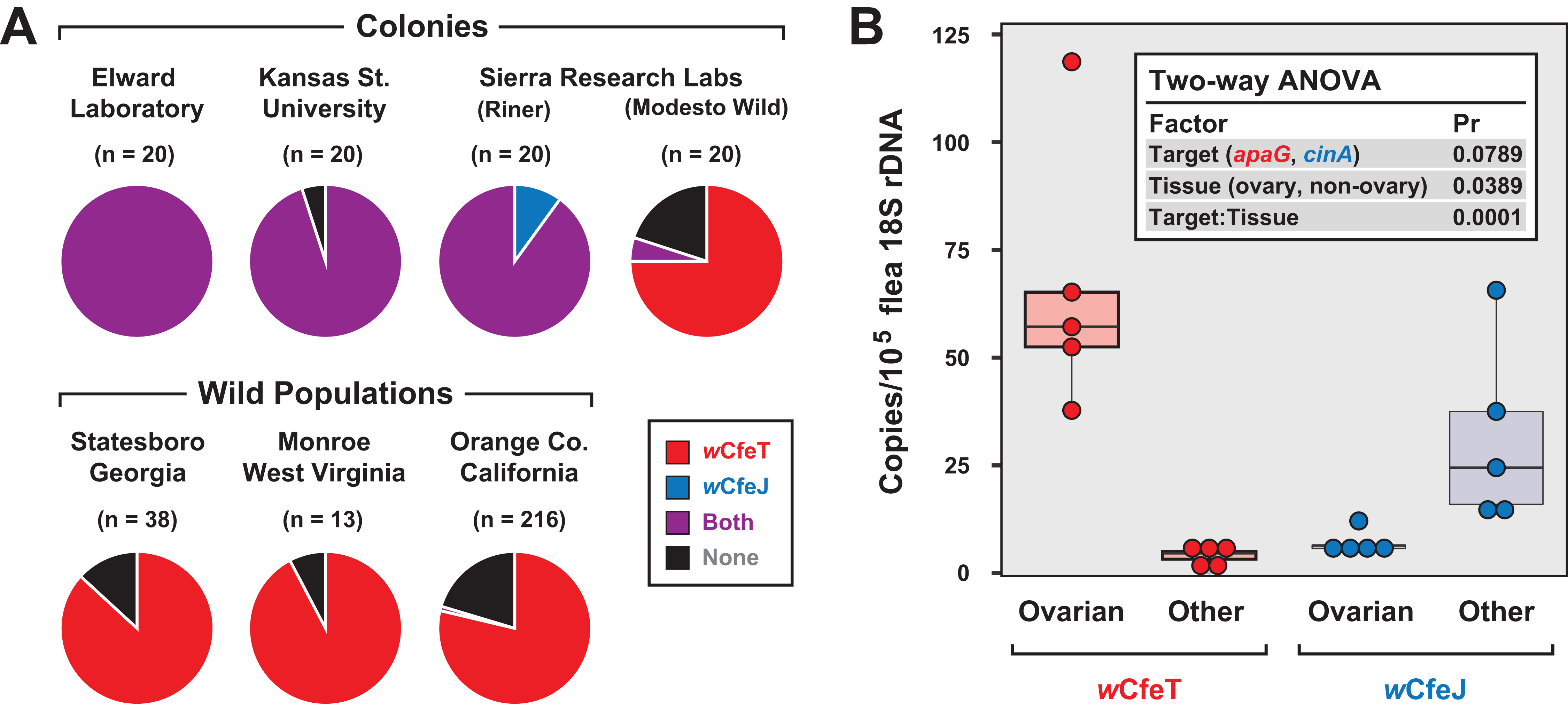{kind=link}
To gain more insight into the nature of the wCfeT-wCfeJ co-infection, we analyzed the distribution of wCfeT and wCfeJ in EL flea tissues (Fig. 5B). Surmising that both Wolbachia strains are maternally transmitted, we dissected out the ovaries of unfed virgin female fleas and compared Wolbachia density in these tissues vs others (primarily midgut). While wCfeT predominantly localizes in flea ovaries, the cif carrier, wCfeJ, mostly localizes to somatic tissues, although minimal co-localization was observed. The significance of this distribution is unclear. wCfeJ may relocate to ovaries upon a mating cue, or perhaps exert CI via some undefined somatic cell mechanism. Transmission of wCfeJ may also be predominantly horizontal via environmental or direct flea contact (per (Pietri, DeBruhl & Sullivan, 2016)). Sampling co-infected fleas at specific timepoints during development, as well as determining wCfeJ tissue localization in the absence of wCfeT infection, will be required to unravel the intertwined transmission dynamics of these Wolbachia strains in cat fleas.
Evolution of Wolbachia mutualism and reproductive parasitism
Order Rickettsiales is a highly diverse group of obligate intracellular bacteria (Gillespie et al., 2012; Driscoll et al., 2013; Darby et al., 2007). A rapid pace of newly identified lineages (Klinges et al., 2019; Castelli et al., 2019; Tashyreva et al., 2018; Kamm et al., 2019; Gruber-Vodicka et al., 2019; Martijn et al., 2015; Schulz et al., 2016; Yurchenko et al., 2018; Schrallhammer et al., 2013; Montagna et al., 2013; Boscaro et al., 2019; Muñoz-Gómez et al., 2019; Mediannikov et al., 2014; Szokoli et al., 2016; Prokopchuk et al., 2019) provides a rich source for evaluating mechanisms of evolution within constraints of the eukaryotic cell. Phage, plasmids, transposons, ICEs, and other insertion sequences are variably present across rickettsial genomes and mobilize numerous genes to offset reductive genome evolution and provide lifestyle altering traits (Gillespie et al., 2012). Our analyses of Wolbachia BOOM and RP genes prompted us to evaluate the recurrent evolution of nutritional-mediated mutualism and reproductive parasitism across diverse rickettsial lineages (Fig. 6).
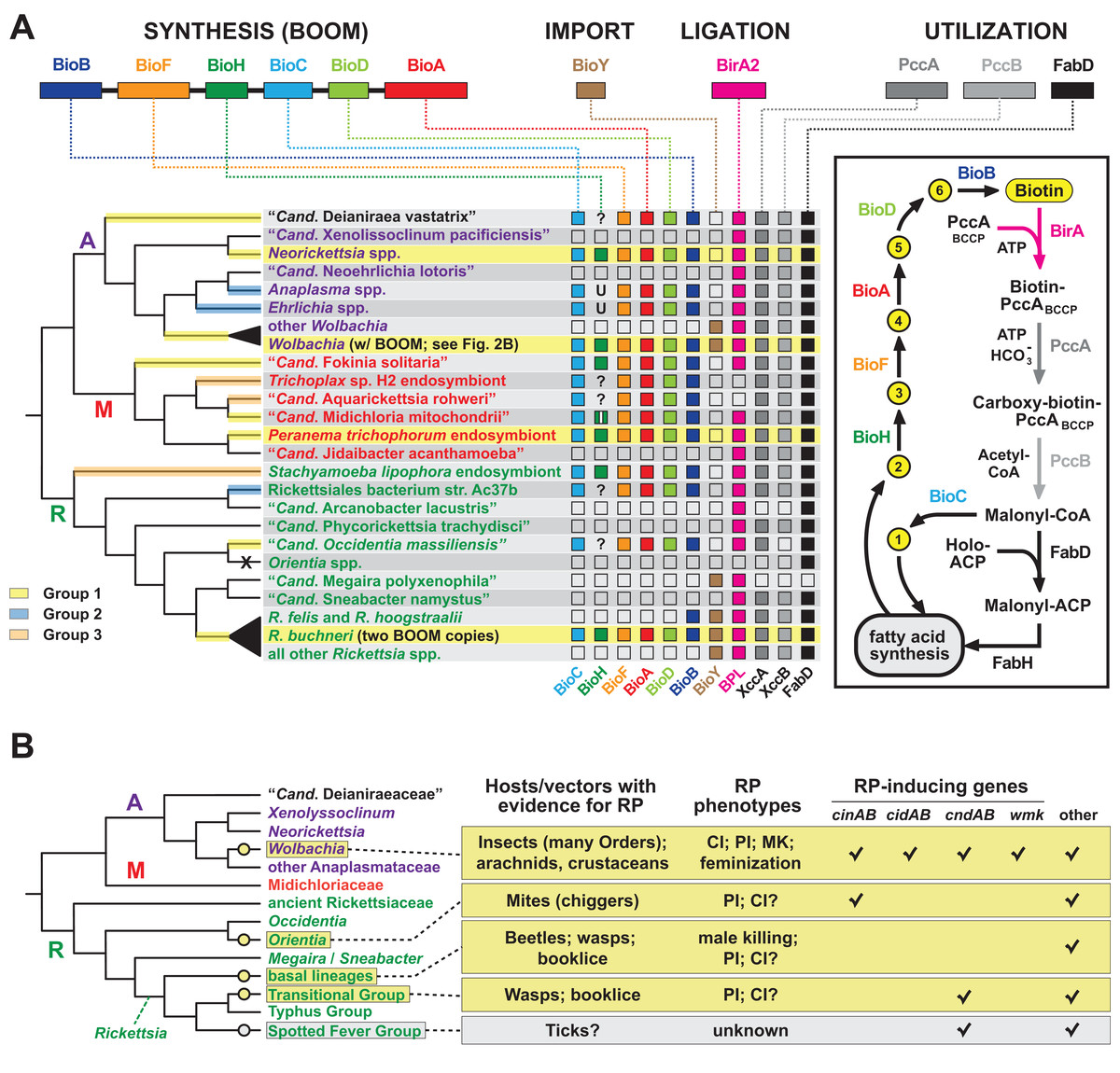
Figure 6: Recurrent evolution of nutritional-mediated mutualism and reproductive parasitism across diverse rickettsial lineages.
(A) Diverse strategies for biotin biosynthesis, acquisition and utilization in Rickettsiales. Top, biotin synthesis genes (BOOM arrangement), the BioY biotin transporter, the biotin ligase BirA, and biotin utilization enzymes (biotin-dependent propionyl-CoA carboxylase complex and FabD) involved in malonyl-CoA synthesis. The distribution of these genes across major rickettsial lineages is summarized, with phylogeny of Rickettsiales drawn as a consensus from recent studies (Driscoll et al., 2017; Castelli et al., 2019; Muñoz-Gómez et al., 2019; Szokoli et al., 2016). The acquisition of biotin synthesis operons is depicted by yellow, blue and orange highlighting over branches (see estimated phylogeny in Fig. 1A). Taxa highlighted yellow indicate the conservation of BOOM gene arrangement (see Fig. S3). Question marks indicate missing BioH enzymes in otherwise complete biotin synthesis pathways. These genomes were scanned for other functionally equivalent methyl esterases and found to contain none (Fig. S4D). Inset highlights the gatekeeper role of BioH in shunting X Pimeloyl-CoA methyl ester (metabolite 2) away from fatty acid biosynthesis for the production of biotin. All other metabolites in the biotin synthesis pathway are described in Fig. 2A. X, complete loss of biotin utilization in O. tsutsugamushi. (B) Phylogenomic analysis of RP in Rickettsiales. Phylogeny estimation is further simplified relative to the tree in panel A. RP is considered an acquired trait, with origins shown as yellow circles. Spotted Fever Group rickettsiae are shown in gray since RP is unknown, though putative RP-inducing genes are present in some species. Information on rickettsial hosts/vectors (Gillespie et al., 2012) and known RP (Gillespie et al., 2018; Beckmann et al., 2019) are compiled from numerous reports. Check marks reflect the compilations of groups of RP-inducing genes from our prior report, or the recent identification of the WO-mediated killing (wmk) gene implicated in Wolbachia male-killing (Perlmutter et al., 2019). Helix-turn-helix domains of certain XRE-family like proteins across Rickettsiales were not considered related to wmk-encoded proteins. “Other” refers to additional proteins presented in our prior report, such as OTU domain-containing proteins and proteins containing the general CndB scaffold (Gillespie et al., 2018).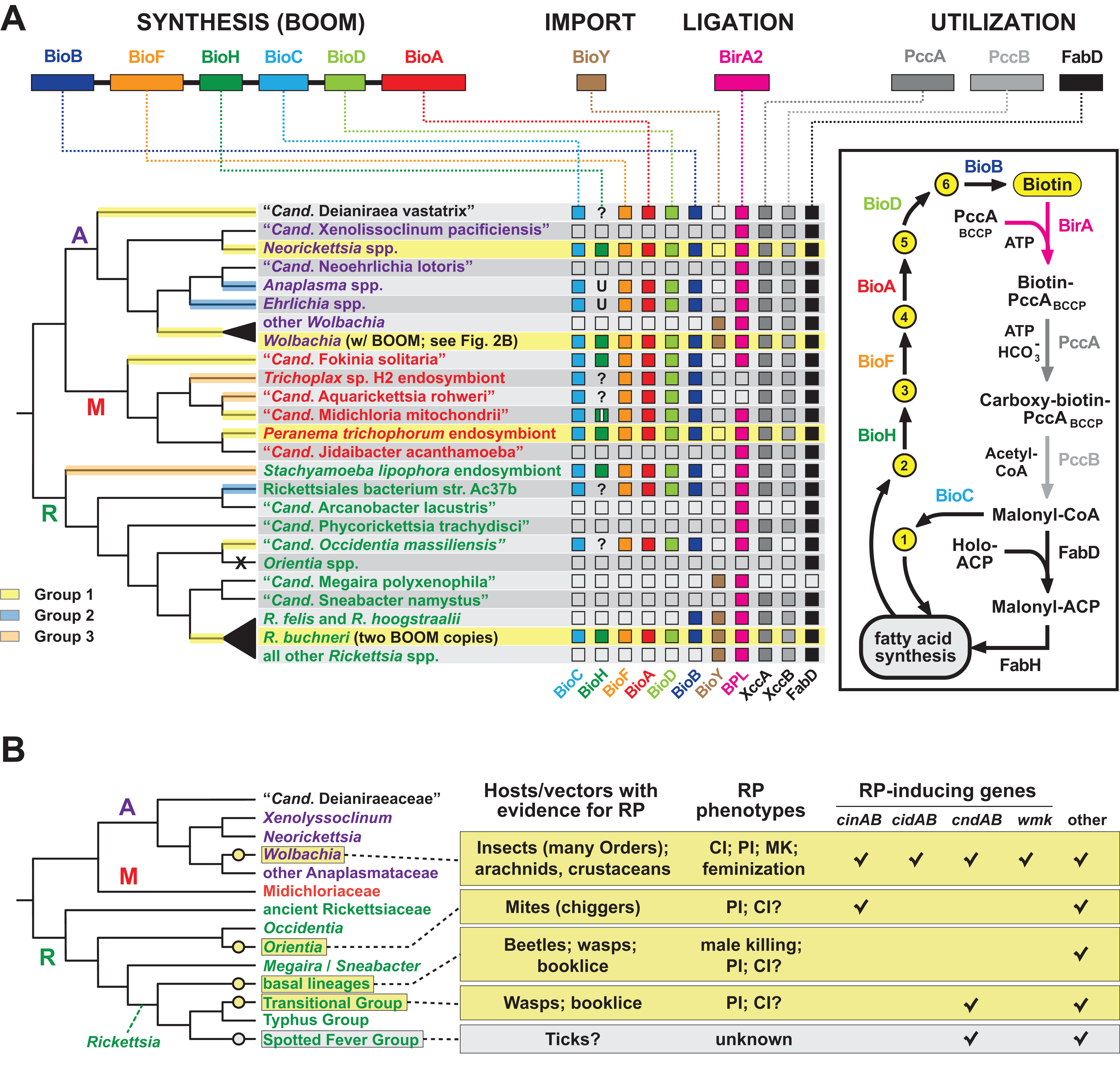{kind=link}
Nutritional-mediated mutualism
Phylogeny estimation supports three independent acquisitions of biotin biosynthesis genes by rickettsial species (Fig. 6A; Fig. S1A). The largest clade (group 1) contains all BOOM-containing species, though only Neorickettsia spp., certain wolbachiae, R. buchneri, and the Peranema trichophorum endosymbiont carry the conserved BOOM gene arrangement (highlighted yellow in Fig. 6A). Other species in this group, as well as Groups 2 and 3, show different gene arrangements indicating recombination and/or possible replacement of specific bio genes through additional LGT (Fig. S3). BOOM and other bio gene clusters have repeatedly lost bioH, which encodes the methyl esterase generating pimeloyl-CoA, the last step in the synthesis of the pimelate moiety of biotin. Bacteria have evolved numerous strategies for catalyzing this reaction (Lin & Cronan, 2011; Shapiro, Chakravartty & Cronan, 2012; Bi et al., 2016; Feng et al., 2014; Zeng et al., 2020; Chow et al., 2018; Shi et al., 2016; Estrada et al., 2017; Sullivan et al., 2001). Interestingly, a novel methyl esterase of Ehrlichia chaffeensis (BioU) was recently shown to complement an E. coli ΔbioH mutant (Hang et al., 2019); however, we determined that BioU orthologs are present in many rickettsial species, including some that harbor a BioH already and others with no bio synthesis genes at all (Figs. S4A–S4C). This indicates that while BioU may generate pimeloyl-CoA in certain rickettsial species—namely, those with complete bio synthesis modules except for BioH—it is likely to have a broad range of functions in bacteria (Fig. S4D). Furthermore, some species lack the genes for both BioH and BioU, indicating that alternative pathways for generating pimeloyl-CoA await discovery.
Analysis of the genomic distribution of biotin synthesis genes, the biotin transporter BioY, the biotin ligase BirA, and the enzymes involved in malonyl-CoA synthesis (biotin-dependent propionyl-CoA carboxylase complex and FabD) revealed several strategies for biotin utilization in Rickettsiales (Fig. 6A). Species that harbor a complete set of bio genes (as discussed above) also contain ligase and utilization enzymes, although (not surprisingly) most do not carry the BioY importer. Genera Wolbachia and Rickettsia largely rely on parasitic scavenging of host biotin to fuel fatty acid biosynthesis: they are dominated by species that lack bio genes but carry the BioY importer, BirA ligase, and utilization enzymes. Two species of Transitional Group Rickettsiae (R. felis and R. hoogstraalii) may be able to utilize imported dethiobiotin as well, as they retain the biotin synthase BioB; alternatively, this may represent the frayed end of a decaying metabolic pathway similar to the glycerol phospholipid and folate biosynthesis pathways in Rickettsiae (Driscoll et al., 2017; Audia, 2012). While characterized as two-component transporters (with BioM/N), BioY subunits alone can efficiently transport biotin into cells (Hebbeln et al., 2007) and also participate with host transporters to relocate cytosolic biotin to vacuoles occupied by pathogens (Fisher et al., 2012). However, certain rickettsial species lacking synthesis genes also lack BioY importers yet retain the ligase and utilization enzymes, indicating the presence of alternative transport mechanisms. Remarkably, Orientia spp. lack all genes involved in biotin synthesis, import, ligation and utilization, exhibiting the ultimate form of parasitism for host-dependent fatty acid synthesis: these Rickettsiae must pilfer malonyl-CoA from the host (Nakayama et al., 2008), using a transport system yet to be described.
The acquisition of BOOM by certain wolbachiae and R. buchneri provides a theoretical framework for how LGT facilitates a transition from metabolite thievery to nutritional-mediated symbiosis. However, it is important to consider that these endoparasites may be utilizing the BOOM primarily for selfish purposes, as almost all bacteria require biotin. For example, Neorickettsia spp. and the deltaproteobacterium Lawsonia intracellularis are pathogens that also harbor the BOOM, suggesting a more complex ecological role surrounding biotin acquisition. If Neorickettsia spp. do indeed actively provision biotin to their trematode and insect vectors, as has been shown for wCle (Nikoh et al., 2014), such host-specific supplementation would necessitate as-yet unidentified genes encoding export factors as well as stringent regulation of the BOOM.
Canonical regulation of biotin synthesis is accomplished via a repressor, fused to the N-terminal domain of the biotin ligase gene, which down-regulates transcription of the bio gene cluster in response to high cellular biotin (Barker & Campbell, 1981; Satiaputra et al., 2016). Interestingly, all rickettsial BirA biotin ligases lack this repressor domain, possibly indicating alternative mechanisms for regulating biotin synthesis and host supplementation by these bacteria. “Candidatus Aquarickettsia rohweri” and the Trichoplax sp. H2 endosymbiont (Midichloriaceae) lack BirA ligases altogether (Fig. 6A), and no other biotin ligases were found in these genomes. Regarding the Trichoplax sp. H2 endosymbiont, our prior analysis of the closely related Rickettsiales endosymbiont of Trichoplax adhaerens (RETA) discovered two biotin-protein ligase N-terminal domain-containing proteins (BPL_N) encoded in the T. adhaerens genome (Driscoll et al., 2013). These host-encoded genes contain introns and eukaryotic regulatory elements yet are strongly supported as rickettsial in origin. Homologs of these T. adhaerens proteins are also present in the Trichoplax sp. H2 genome (Kamm et al., 2018) (NCBI acc. nos. RDD42697 and RDD42436), supporting their cross-domain LGT. This raises the possibility for a metabolic interdependence involving biotin synthesis and utilization in placozoans and their rickettsial symbionts, similar to the mosaic metabolic networks underpinning nutritional symbioses in other systems (Bublitz et al., 2019; McCutcheon, 2010; Wilson & Duncan, 2015). For example, the nested tripartite symbiosis between Planococcus citri mealybugs, Tremblaya princeps, and Moranella endobia shows evidence for LGT of several bio genes to the mealybug genome (Husnik et al., 2013); these genes share highest similarity to Wolbachia BOOM counterparts. This is reminiscent of the pea aphid-Buchnera symbiosis, wherein several rickettsial LGTs have been implicated in facilitating metabolic interdependence between host and symbiont (Nikoh et al., 2010). For rickettsial endosymbionts of placozoans, host acquisition of BPL_N genes (among others (Driscoll et al., 2013)) may have established an inextricable obligatory relationship (McCutcheon, 2016; McCutcheon, Boyd & Dale, 2019) and may hint at the evolutionary fate of other BOOM-containing rickettsiae. However, while our analyses of the BOOM and other bio genes in rickettsial genomes are suggestive, in the absence of experimental validation we urge caution against implicating these genes as factors underpinning nutritional-mediated mutualism.
Reproductive parasitism
Among other bacterial reproductive parasites of arthropods (e.g., species of Cardinium, Arsenophonus and Spiroplasma) wolbachiae are perhaps the best-studied for their roles in RP induction (Hurst & Frost, 2015). Yet within the Rickettsiales, wolbachiae do not walk alone in inducing RP in their hosts, as three other lineages contain species known to induce RP phenotypes (Orientia tsutsugamushi, basal Rickettsia species and Transitional Group Rickettsiae). Inspection of genomic data indicates SFG Rickettsiae may also harbor reproductive parasites (Fig. 6B). These analyses reveal four discrete molecular determinants for RP induction that have evolved throughout rickettsial evolution (cinAB, cidAB, cndAB, wmk), though others likely exist based on previous description of numerous gene models sharing commonalities across toxins and antidote architectures (Gillespie et al., 2018). We posit at least four independent gains of RP in the Rickettsiaceae have occurred, coupled with widespread RP seen in wolbachiae (Fig. 6B). This speculative interpretation is more parsimonious than treating RP as an ancestral rickettsial trait, given that RP-inducing genes strongly associate with mobile genetic elements (e.g., WO prophage in Wolbachia strains, RAGE ICEs and plasmids in Rickettsia species). Additionally, were RP an ancestral rickettsial trait, one would expect to see cifs in Rickettsia genomes more frequently than what is observed based on analysis of available genomes (Gillespie et al., 2018).
In contrast to rickettsiae, RP-inducing genes are found in greater numbers and diversity in wolbachiae, undoubtedly due to their strong association with EAMs of WO prophage genomes and mobile elements (Bordenstein & Bordenstein, 2016; Lindsey et al., 2018). In our previous work, we uncovered a link between the characterized CI-inducing CinA/B and CidA/B operons of wolbachiae and a putative TA operon encoded on the pLbAR plasmid of Rickettsia felis str. LSU-Lb, an obligate endosymbiont of the booklouse Liposcelis bostrychophila (Gillespie et al., 2015). We posited that this TA operon induces parthenogenesis in the booklouse: pLbAR is unknown in other strains of R. felis which do not induce parthenogenesis, in booklice or other arthropods (Gillespie et al., 2015), and booklice lacking R. felis str. LSU-Lb reproduce sexually (Yang et al., 2015; Mockford & Krushelnycky, 2008). Aside from being a CndB toxin (harboring both the NUC and DUB domains of wolbachiae CinB and CidB toxins, respectively) the large pLbAR toxin (3,241 aa) was found to carry several other domains present in other Wolbachia proteins, most of which are also EAM-associated. These domains, together with the large central domain (LCD) of the pLbAR toxin, were utilized to identify an extraordinarily diverse array of candidate RP-inducing toxins in a wide-range of intracellular bacteria, some of which are known reproductive parasites (Gillespie et al., 2018). Linked within the intracellular mobilome, we consider this vast family of proteins a source for the recurrent evolution of RP in invertebrate-borne bacteria.
Aside from CI induction by CinA/B and CidA/B operons (Chen et al., 2019; Beckmann et al., 2019; LePage et al., 2017; Beckmann, Ronau & Hochstrasser, 2017), virtually nothing is known about the functions of related candidate RP-inducing genes. The function of the CinA/B-like operon of O. tsutsugamushi is unclear. O. tsutsugamushi is the pathogen causing scrub typhus and is capable of inducing parthenogenesis in Leptotrombidium mites (Roberts, Rapmund & Cadigan, 1977; Takahashi et al., 1997). Given this fact, exploration of its corresponding CinA/B module is warranted. Though the connection to RP in mites is unclear given that CinA/B only occurs in the Boryong strain; a myriad of other proteins encoding the pLbAR LCD are amplified in all Orientia genomes. Genome sequences are needed to determine toxin profiles for Rickettsia species either in basal lineages (Perotti et al., 2006; Gualtieri et al., 2017; Giorgini et al., 2010) or Transitional Group rickettsiae (Perotti et al., 2006; Hagimori et al., 2006; Nugnes et al., 2015) that induce parthenogenesis in eulophid wasps or trogiid booklice. Genome sequences are also lacking for Rickettsia species from basal lineages known to induce male-killing in buprestid and ladybird beetles (Werren et al., 1994; Von der Schulenburg et al., 2001; Lawson et al., 2001; Majerus & Majerus, 2010). We previously mined the genome of the male killer Rickettsia parasite of the ladybird beetle Adalia bipunctata (Murray et al., 2016) and found a toxin highly similar in architecture to the Spiroplasma male killer toxin (Harumoto & Lemaitre, 2018). Both toxins carry an ovarian tumor (OTU) cysteine protease domain (Makarova, Aravind & Koonin, 2000), which is a deubiquitylating enzyme. Further searching with the OTU domain resulted in a hodgepodge of chimeric toxin architectures from bacteria known to induce other RP phenotypes, such as parthenogenesis (wFol (Faddeeva-Vakhrusheva et al., 2017)) and feminization (wVulC (Badawi et al., 2018)). Thus, it is clear that additional high-quality (closed) genomes are sorely needed to correlate RP-inducing protein domains and RP phenotypes. New correlations will lead to downstream functional analyses of target’s abilities to induce RP.
A large number of candidate proteins from the pLbAR-derived assemblage occur in species that are not known as reproductive parasites. These include CndB toxins, NUC- and DUB-domain containing proteins that are much larger than characterized CinB and CidB toxins, diverse OTU-domain containing toxins, and especially proteins harboring the LCD that may or may not exhibit toxin-like profiles (Gillespie et al., 2018). Some of these candidate toxins appear to have cognate antidotes, and some genomes carry antidotes only, highlighting the complex diversity of this biological function. Recently, an EAM-encoded transcriptional factor in wMel termed WO-mediated killing (wmk) was shown to induce male-specific lethality during early embryogenesis when expressed transgenically in fruit flies (Perlmutter et al., 2019). This male-killer gene was not detected as part of our pLbAR-derived pool, indicating that RP-inducing gene arsenals are probably much larger than have been described thus far, particularly considering that cin, cid and cnd loci are not present in the genomes of Cardinium and Arsenophonus reproductive parasites (Gillespie et al., 2018; Beckmann et al., 2019; Gebiola et al., 2017) for which the mechanisms for RP induction remain unknown (Siozios et al., 2019; Duron et al., 2008).
Candidate RP-inducing toxins with similar features to the pLbAR toxin (large, modular, and associated with mobile genetic elements) are found in several Wolbachia genomes (e.g., wFol, wStr, wDacB, wVulC, wCon, wCed, and wCauA). They are often in conjunction with other cin and/or cid loci, yet not always associated with WO prophage. Consistent with prior reports identifying LGT between Rickettsia and Wolbachia genomes (Gillespie et al., 2012, 2015; Ishmael et al., 2009; Baldridge et al., 2016), similar toxins in Rickettsia genomes (R. felis str. LSU-Lb, R. gravesii, male killer of Adalia bipunctata) indicate a dynamic platform for disseminating RP-inducing genes across diverse rickettsial species that includes Rickettsia plasmids and WO prophage. Growing numbers of identified Rickettsia plasmids (Gillespie et al., 2015), proliferative RAGE ICEs in Rickettsia and Orientia genomes (Gillespie et al., 2012; Hagen et al., 2018; Nakayama et al., 2008; Batty et al., 2018; Cho et al., 2007; Nakayama et al., 2010), and the recent discovery of the first Wolbachia plasmid (Reveillaud et al., 2019), attest to a highly dynamic mobilome not previously realized for obligate intracellular bacteria. The proposed fission of a CndB-like toxin into a wCfeJ CinB toxin, described in the current study (Fig. 3D), suggests larger toxins undergo streamlining into discrete inducers of CI or other RP phenotypes. As with the evolution of biotin utilization, Rickettsiales provides a framework to study the nature of RP in a diverse array of bacteria that enjoy different relationships with their invertebrate and vertebrate hosts.
Conclusion
The remarkable opposing features of wCfeT and wCfeJ offer testable hypotheses for contrasting host relationships (nutritional symbiosis vs RP, respectively). They also provide a framework to study the dynamics of Wolbachia co-infections, both in colonies and wild populations. The wCfeT and wCfeJ genomes yield lessons on the role of LGT in shaping host associations, highlighting the ephemeral nature of nutritional-based mutualism and RP over a general rickettsial platform of obligate intracellular parasitism. Host genome sequences provide clues to on-going mutualism and historical RP, and our hypothesis for host CI-gene capture leading to suppression of RP illuminates possible limitations for deployment of Wolbachia reproductive parasites to combat arthropod transmission of human pathogens. In summary, our robust characterization of two divergent strains of C. felis-associated wolbachiae, as well as the C. felis genome itself (Driscoll et al., 2020), provides essential information for determining the impact of these bacteria on cat flea biology and pathogen vectoral dynamics.
Supplemental Information
Phylogeny estimations of biotin synthesis enzymes and EamA transporters.
(A) Complete phylogeny estimation of BOOM and other bio gene sets from diverse bacteria. Tree was estimated from the concatenation of six bio enzymes (BioC, BioH, BioF, BioA, BioD, and BioB) or subsets in certain cases (see Table S2 for all sequence information and Materials and Methods for details on dataset processing and tree estimation). Branch support was assessed with 1,000 pseudo-replications. Final ML optimization likelihood was -127140.955066. In taxon color scheme, [R] denotes Rickettsiales, with Holosporaceae [H] as a revised family of Rhodospirillales (101). The families of Rickettsiales are similarly noted: Anaplasmataceae [A], Midichloriaceae [M], Rickettsiaceae [R], and “Candidatus Deianiaeaceae” [D], with the latter considered provisional (91). (B) Estimated phylogeny of EamA transporters. See Materials and Methods for details on dataset processing and tree estimation. Branch support was assessed with 1,000 pseudo-replications. Final ML optimization likelihood was -23058.797227. Taxon color scheme as described in panel A.
In silico characterization of the w CfeT prophage genome.
Schema shows the comparison of the wCfeT and WOVitA1 prophage genomes, with gene descriptions following prior studies (26, 30, 148, 183). wCfeT genes within the typical Eukaryotic Association Module (EAM) were used as queries in Blastp searches against the NCBI nr protein database, with summary statistics provided for top subjects (dashed box).
Synteny analysis of biotin synthesis genes in Rickettsiales genomes.
Rickettsiales taxa with at least 5 of 6 biotin synthesis genes (n=28) were analyzed for co-linearity of their biotin genes. Ortholog groups (n=2,527) were constructed from these genomes as described for Fig. 1 (see Materials and Methods) and OGs containing biotin synthesis genes identified. Genome locations were retrieved from NCBI in gene file format (gff) and used to order the orthologs in each genome. For unclosed genomes, all contigs were included in OG construction; only a single genome (Candidatus Aquarickettsia rohweri) contained biotin synthesis genes on multiple contigs. Biotin genes are colored according to the ideal BOOM shown at the top. Black boxes indicate stretches of unrelated genes. Taxa are grouped into 3 classes according to the synteny of biotin genes in their genomes: Class I taxa demonstrate completely conserved gene order; Class II taxa contain only a single gene out of order (Class IIA: BioA; Class IIB: BioB); Class III taxa exhibit little or no conservation of gene order. All blocks are anchored on BioB for ease of comparison, except Class IIB which are anchored on BioF instead.
Bioinformatics analysis of BioU and related methyl esterases.
(A) Alignment of Ehrlichia chaffeensis and Anaplasma centrale BioU proteins with orthologs from other Rickettsiales species. BioU orthologs were retrieved from Blastp searches against the NCBI Rickettsiales databse using E. chaffeensis str. Sapulpa BioU (EAM85518.1) as a query. The catalytic residues (Ser–Asp–His) characteristic of methyl esterases are highlighted yellow (184). Sequences were aligned with MUSCLE v3.8.31 (169) using default parameters. NCBI protein accession numbers are listed in panel C. (B) Percent identity matrix calculated from the alignment shown in panel A. (C) Estimated phylogeny of proteins shown in panel A. Two alphaproteobacterial outgroups and two hymenopteran mitochondrial methyl esterases are included. Phylogeny was estimated using RAxML v8.2.4 (171) under the gamma model of rate heterogeneity and estimation of the proportion of invariant sites. Branch support was assessed with 1,000 pseudo-replications. Final ML optimization likelihood was -5448.767799. (D) Analysis of eight rickettsial genomes that contain biotin synthesis genes except for the BioH methyl esterase. Blastp searches against the rickettsial genomes were conducted using the following queries, all of which have been shown to orthogonally substitute for BioH: BioV of Helicobacter sp. L8b (TSA87024.1); BioG of Haemophilus influenzae str. CGSHiCZ412602 (AIB45959.1); BtsA of Moraxella catarrhalis str. CCUG 353 (KZR94829.1); BioJ of Francisella marina str. E103-15 (QEO56965.1); BioZ of Mesorhizobium japonicum str. R7A (AAG47795.1); EstN1 of Nitrososphaera viennensis str. EN76 (AIC14436.1); BioK of Prochlorococcus marinus str. SS51 (KGG31655.1).