
Comparative plastomics of Amaryllidaceae: inverted repeat expansion and the degradation of the ndh genes in Strumaria truncata Jacq.
- Published
- Accepted
- Received
- Academic Editor
- Alison Nazareno
- Subject Areas
- Evolutionary Studies, Molecular Biology, Plant Science
- Keywords
- Amaryllidaceae, IR expansion, ndh loss, Strumaria
- Copyright
- © 2021 Könyves et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2021. Comparative plastomics of Amaryllidaceae: inverted repeat expansion and the degradation of the ndh genes in Strumaria truncata Jacq. PeerJ 9:e12400 https://doi.org/10.7717/peerj.12400
Abstract
Amaryllidaceae is a widespread and distinctive plant family contributing both food and ornamental plants. Here we present an initial survey of plastomes across the family and report on both structural rearrangements and gene losses. Most plastomes in the family are of similar gene arrangement and content however some taxa have shown gains in plastome length while in several taxa there is evidence of gene loss. Strumaria truncata shows a substantial loss of ndh family genes while three other taxa show loss of cemA, which has been reported only rarely. Our sparse sampling of the family has detected sufficient variation to suggest further sampling across the family could be a rich source of new information on plastome variation and evolution.
Introduction
The plastid genome, or plastome, in land plants is generally conserved in length, structure, and gene content (Wicke et al., 2011). Typical flowering plant plastomes range from 120 to 160 kb, contain 100–120 unique genes, and have a quadripartite structure of two single copy regions (LSC and SSC) separated by two copies of the inverted repeat (IRs) (Jansen & Ruhlman, 2012; Smith & Keeling, 2015). However, exceptions to all of these features have been found. The greatly reduced plastomes of Pilostyles range from 11–15 kb and contain only seven functioning genes (Arias-Agudelo et al., 2019). In contrast the plastome of Pelargonium × hortorum has expanded to 218 kb including unusually long inverted repeats (Chumley et al., 2006). Plastomes deviating from the quadripartite structure have also been reported, either without one copy of the IR (Wojciechowski et al., 2000; Sanderson et al., 2015) or incorporating the entire SSC into the inverted repeats (Sinn et al., 2018).
Rearrangements are often associated with increased repeat content in plastid genomes (Chumley et al., 2006; Haberle et al., 2008; Guisinger et al., 2011). One of the most commonly reported plastome rearrangements is the expansion or contraction of the inverted repeats. Palmer et al. (1985) and Yamada (1991) hypothesised that intramolecular recombination at short inverted repeats located within and around the main IR resulted in the expansion of the Chlamydomonas reinhardtii and Chlorella ellipsoidea inverted repeats, respectively, while Aii et al. (1997) proposed that recombination at forward repeats within ycf1 lead to an inversion, and the IR expansion in buckwheat (Fagopyrum sp.). However, in the absence of short repeats in Nicotiana, Goulding et al. (1996) established two different mechanisms for both short, <100 bp, and long, over 10 kb, IR expansions: short expansions are the result of gene conversion after heteroduplex formation via Holliday junctions, and long expansions are the result of a double-strand break (DSB) in one of the IRs followed by strand invasion and repair against the other IR within the same plastome unit that progresses through the junction incorporating single-copy regions into the inverted repeats. A similar DSB repair process, but via homologous recombination at imperfect nonallelic repeats between different plastome units, resulted in the reestablishment of the IR in Medicago (Choi, Jansen & Ruhlman, 2019). Zhu et al. (2016) attributed the IR expansions in several dicot lineages to the gene conversion mechanism. Furthermore, Wang et al. (2008) proposed that the DSB mechanism of Goulding et al. (1996) can account for shorter expansions as well, such as the development of the monocot-type IR/LSC junction by incorporating rps19 and trnH into the IR.
Various gene losses have occurred during the evolution of the angiosperm plastome (Raubeson & Jansen, 2005; Jansen et al., 2007); a commonly reported example is the loss of ndh genes (Song et al., 2017; Silva et al., 2018; Nevill et al., 2019). The loss of ndh genes is strongly associated with a change in trophic conditions (Wicke et al., 2013; Graham, Lam & Merckx, 2017); it represents the first step in plastome degradation in heterotrophic plants (Martín & Sabater, 2010; Barrett & Davis, 2012). Apart from plant lineages less reliant on photosynthesis, multiple ndh gene losses have also been reported in fully photosynthetic lineages for example in aquatic/semi-aquatic plants (Iles, Smith & Graham, 2013; Peredo, King & Les, 2013; Folk et al., 2020), gymnosperms (Wakasugi et al., 1994; McCoy et al., 2008), Orchidaceae (Kim et al., 2015; Roma et al., 2018), and Cactaceae (Sanderson et al., 2015; Köhler et al., 2020).
Amaryllidaceae J. St.-Hil. (Saint-Hilaire, 1805) is a cosmopolitan family of bulbous geophytes and rhizomatous perennials in Asparagales (Meerow, 2000) comprising approximately 90 genera, and over 1,700 species (Meerow, Gardner & Nakamura, 2020), in three subfamilies: Amaryllidoideae, Allioideae, and Agapanthoideae, all of which share an umbellate inflorescence. This family of petaloid monocots contains many horticulturally important genera, including: Agapanthus, Allium, Amaryllis, Clivia, Galanthus, Hippeastrum, Narcissus, and Nerine (Heywood et al., 2007). Amaryllidoideae, the most diverse subfamily with c. 75 genera (Meerow, Gardner & Nakamura, 2020), has a complex evolutionary history, including hybridisation (García et al., 2017; Marques et al., 2017; Meerow, Gardner & Nakamura, 2020), and morphological convergence (Meerow, 2010). Arising from our research, in horticulturally important Amaryllidaceae genera (Könyves et al., 2018; Könyves et al., 2019a; David & Könyves, 2019), here we report the sequencing and assembly of five Amaryllidoideae species and compare our assemblies with available Amaryllidaceae plastomes from GenBank. This will broaden the knowledge, and reporting, of plastome structural variation in Amaryllidaceae.
Materials & Methods
Fresh leaf material was collected from five Amaryllidoideae species at RHS Garden Wisley, UK or from a private collection (Table 1). Our sampling was fundamentally opportunistic, including Amaryllidoideae species that were growing at the time of collection, and related to other projects we were working on (Könyves et al., 2018; Könyves et al., 2019a; David & Könyves, 2019). It did, however, broaden the generic level sampling of the subfamily because these genera were chosen to act as outgroups to other, generic-level studies. Herbarium voucher specimens were deposited at WSY. Total genomic DNA was extracted using the QIAGEN DNeasy Plant Mini Kit (QIAGEN, Manchester, UK). Library development and 150 bp PE (paired-end) sequencing on an Illumina HiSeq 4000 lane was done by the Oxford Genomics Centre (Oxford, UK). The plastomes were assembled with Fast-Plast v1.2.6 (McKain & Wilson, 2017) and NOVOPlasty v2.7.0 (Dierckxsens, Mardulyn & Smits, 2017). Fast-Plast assemblies were run with a total of 5M, 10M, 20M reads (i.e., 2.5M, 5M, 10M PE reads) and with all available reads. Reads were trimmed to remove NEB-PE adapter sequences. Bowtie reference indices were built with the published Narcissus poeticus plastome (MH706763). For the NOVOPlasty assemblies, adapters were trimmed with Trimmomatic v0.36 (Bolger, Lohse & Usadel, 2014), using the same adapter sequences. An ndhF sequence of Na. poeticus (KT124416) was used as the starting seed and memory was limited to 8 Gb. All other parameters were unchanged. The Strumaria truncata NOVOPlasty assembly failed with the ndhF seed, a trnK/matK sequence of Na. poeticus (KC238498) was used instead. Among those assemblies that did not produce consistent results across the different assembly strategies, the large single copy (LSC), the small single copy (SSC), and two inverted repeat (IR) regions were identified in the final Fast-Plast contig and NOVOPlasty assemblies, and the circular plastome was assembled by hand using Geneious v11.1.5 (http://www.geneious.com; Kearse et al., 2012). The junctions of the inverted repeats and the ndh gene sequences in the S. truncata plastome assembly were confirmed by Sanger sequencing using the primers and PCR protocols detailed in Tables S1 and S2. Coverage analysis of the finished plastomes were done in Fast-Plast. The complete plastomes were annotated by transferring equivalent annotations from the Na. poeticus plastome using Geneious v11.1.5. Gene and exon boundaries were corrected by hand when necessary.
| Species | GenBank acc. no. | Source |
|---|---|---|
| Acis autumnalis var. oporantha (Jord. & Fourr.) Lledó, A.P.Davis & M.B.Crespo | MN539611 | This study (WSY0153095) |
| Lapiedra martinezii Lag. | MN539612 | This study (WSY0153095) |
| Nerine sarniensis (L.) Herb. | MN539613 | This study (WSY0153096) |
| Pancratium maritimum L. | MN539614 | This study (WSY0153097) |
| Strumaria truncata Jacq. | MN539615 | This study (WSY0153098) |
| Clivia miniata (Lindl.) Verschaff. | MN857162 | GenBank (Wang et al., 2020) |
| Hippeastrum rutilum (Ker Gawl.) Herb. | MT133568 | GenBank (Huang, 2020) |
| Leucojum aestivum L. | MH422130 | GenBank (Li et al., 2018) |
| Lycoris radiata (L’Hér.) Herb. | MN158120 | GenBank (Zhang et al., 2019) |
| Lycoris squamigera Maxim. | MH118290 | GenBank (Jin et al., 2018) |
| Narcissus poeticus L. | MH706763 | GenBank (Könyves et al., 2018) |
| Allium altaicum Pall. | MH159130 | GenBank ( Filyushin et al., 2018) |
| Allium fistulosum L. | MH926357 | GenBank (Yusupov et al., 2019) |
| Allium praemixtum Vved. | MK411817 | GenBank (Yusupov et al., 2021) |
| Agapanthus coddii F.M.Leight. | KX790363 | GenBank (unpublished) |
| Hesperoyucca whipplei (Torr.) Trel. | KX931459 | GenBank (McKain et al., 2016) |
| Hemerocallis fulva (L.) L. | MG914655 | GenBank (Lee et al., 2019) |
The plastomes constructed in this study were combined with ten plastomes representing all three subfamilies of Amaryllidaceae, and a further two Asparagales (Asparagaceae and Asphodelaceae) plastomes from GenBank (Table 1). Sixty-seven coding genes (CDS) that are shared between all samples were extracted from the whole plastomes and aligned with the MUSCLE algorithm v3.8.425 using the default parameters in Geneious Prime 2020.0.5 (http://www.geneious.com). Prior to alignment, annotations of GenBank sequences were amended to correct reading frames, where necessary (changes listed in Table S3). The alignments of the 67 CDS were concatenated into a matrix of 58,230 bp. A maximum likelihood estimate of phylogeny was conducted with RAxML v8.2.11 (Stamatakis, 2014) within Geneious Prime using 1000 bootstrap replicates according to the best-fit model of evolution, GTR+I+G, identified by jModelTest 2 (Guindon & Gascuel, 2003; Darriba et al., 2012).
The inverted repeat boundaries for all samples were identified using the Repeat Finder v1.0.1 plugin in Geneious Prime, with default settings. We classified the plastomes into three groups (Type A, B, C) based on the 5′ portion of ycf1 present in IRA, i.e., the structure of the IRA-SSC junction (JSA), to identify IR expansion events. We did not identify any inversions in the plastomes therefore we tested whether the IRA-SSC expansions could have happened through recombination at short inverted repeats present, i.e., the mechanism proposed by Palmer et al. (1985) and Yamada (1991), or through short forward repeats which could mediate recombination as mentioned by Choi, Jansen & Ruhlman (2019). We searched for short repeats present in both target regions in each species of interest, detailed below, using the Repeat Finder v1.0.1 plugin in Geneious Prime with a minimum of 16 bp length and allowing 10% mismatch. We chose the minimum length and allowed for mismatches as Staub & Maliga (1994) showed evidence of recombination at such repeats in plastids. In Na. poeticus and Pancratium maritimum (plastome Type B) we screened the region 500–1,500 bp from the 5′ end of ycf1 (the position where JSA in Type A plastomes occurs, see Figs. 1 and 2) and the 500 bp either side of JSA in both species. Repeat Finder did not identify any repeats in Na. poeticus so we manually checked the target regions for those homologous with P. maritimum to see if this lack is due to our strict search settings. To further investigate the loss of the ndh genes in S. truncata, we screened for the repeats in Nerine sarniensis as well, 500 bp either side of JSA and within ndhH, where JSA in S. truncata is found. The inverted repeat in S. truncata contains a 45 bp region downstream of the ndhH pseudogene that is absent in other taxa. To identify where this 45 bp region originated we aligned a portion of the S. truncata plastome, spanning from trnN in IRB to trnN in IRA, against the same portion of the Ne. sarniensis assembly using the MUSCLE algorithm with the default parameters in Geneious Prime. We identified tandem repeats using Phobos v3.3.12 (Mayer, 2006–2010), within Geneious Prime, following Joyce et al. (2019), by restricting the search to perfect repeats between 2 and 1,000 bp long, with the “remove hidden repeats” setting enabled.
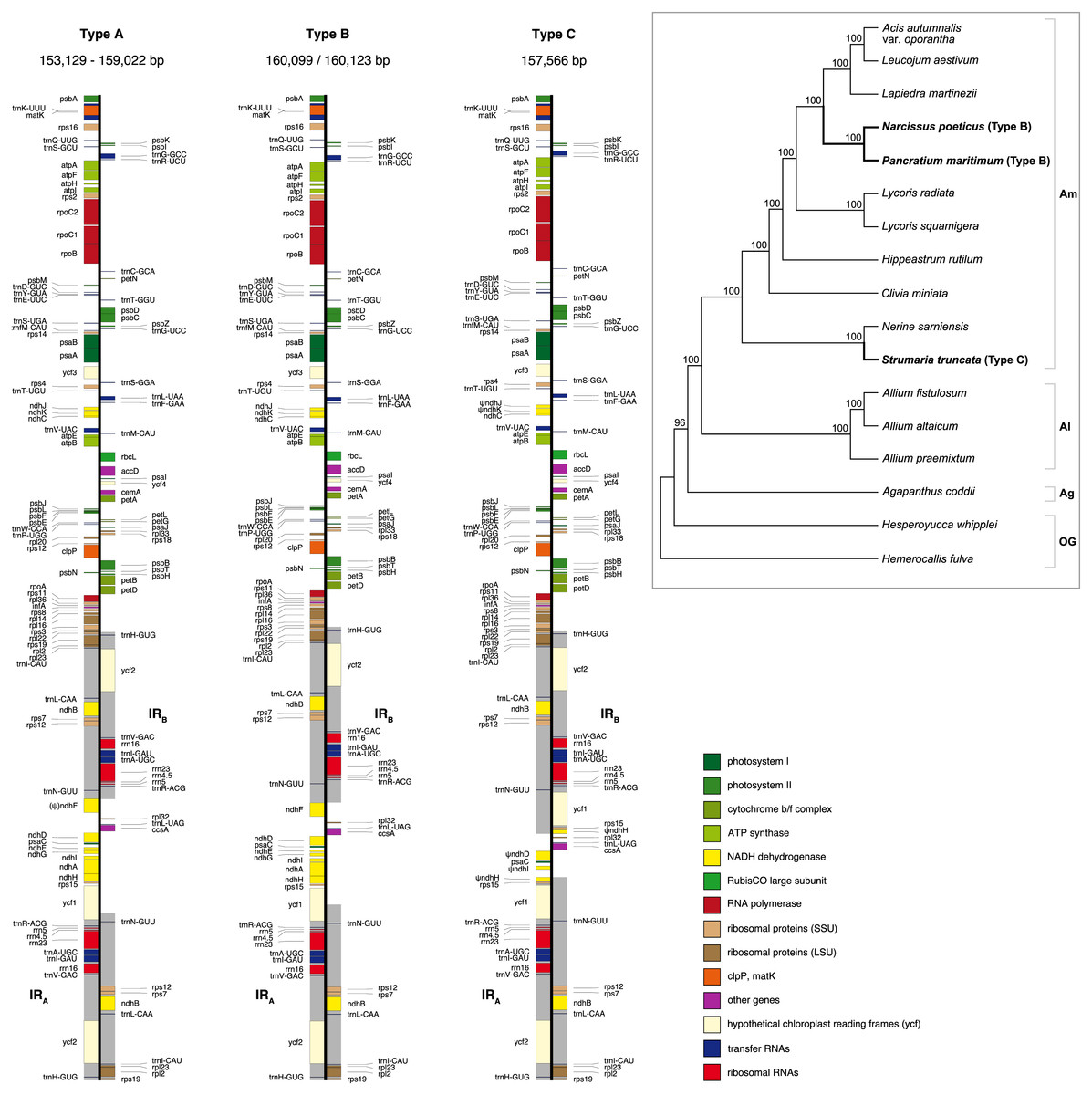
Figure 1: Maps of the three types of plastomes characterised by the 5′ portion of ycf1 present in IRA.
Gray shading highlights the IR regions. Genes are coloured according to functional groups shown in the legend. Inset panel shows the relationship between the sampled species based on the RAxML analysis of 67 coding sequences. Bootstrap support values are shown at nodes. Type B and C plastomes are highlighted in bold, all other samples had Type A plastomes. Amaryllidaceae subfamilies are indicated on the right: Am, Amaryllidoideae; Al, Alliodideae; Ag, Agapanthodieae; OG, Outgroups.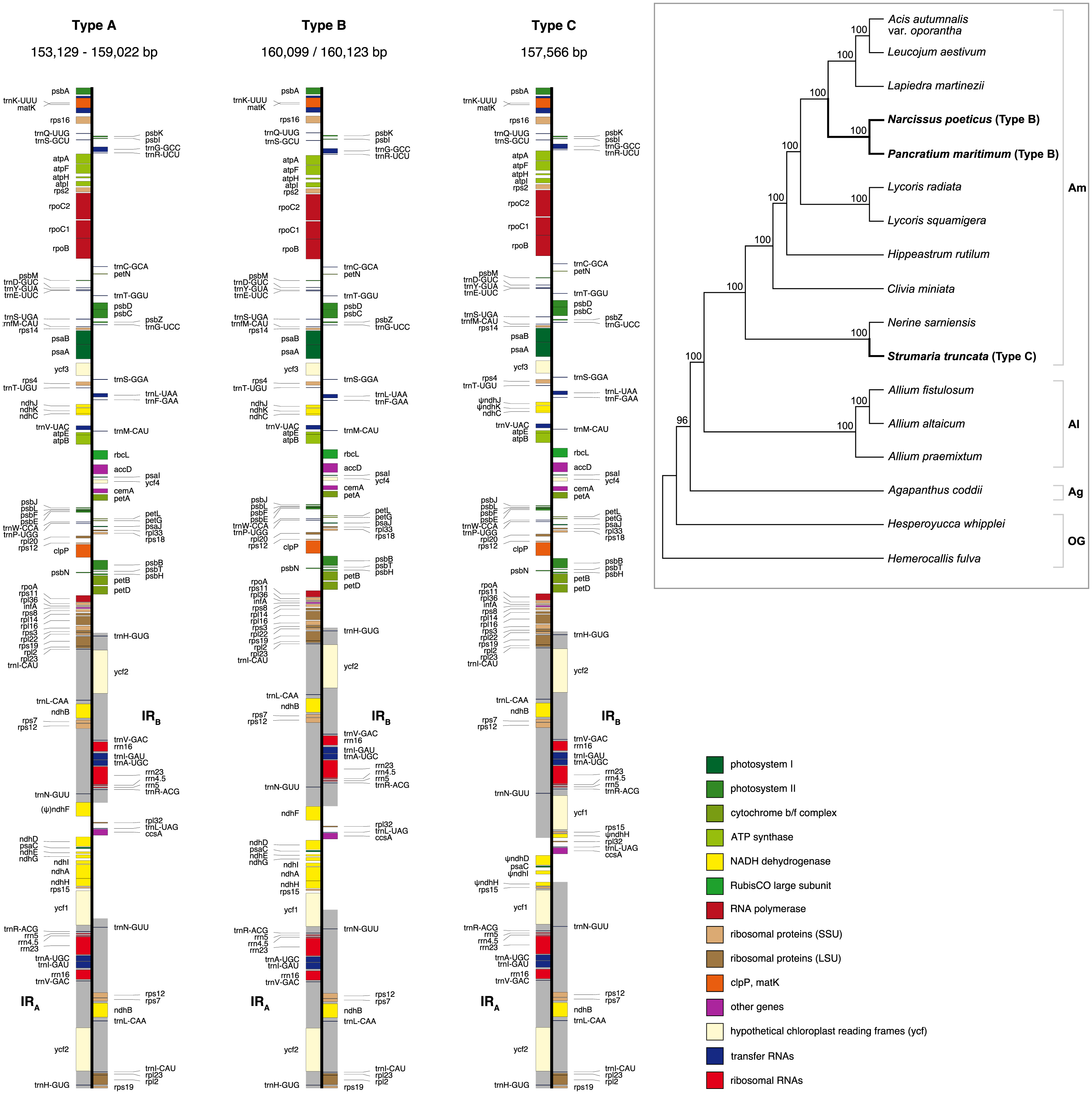{kind=link}
Figure 2: Structure of the junctions (JSA, JSB) between the inverted repeats (IRA, IRB) and small single copy (SSC) in the three plastome types identified in this study.
The portion of genes included in the IRs and SSC is indicated. Gene order, direction of transcription, and colour code of each gene correspond to Fig. 1. ψ indicates pseudogenes.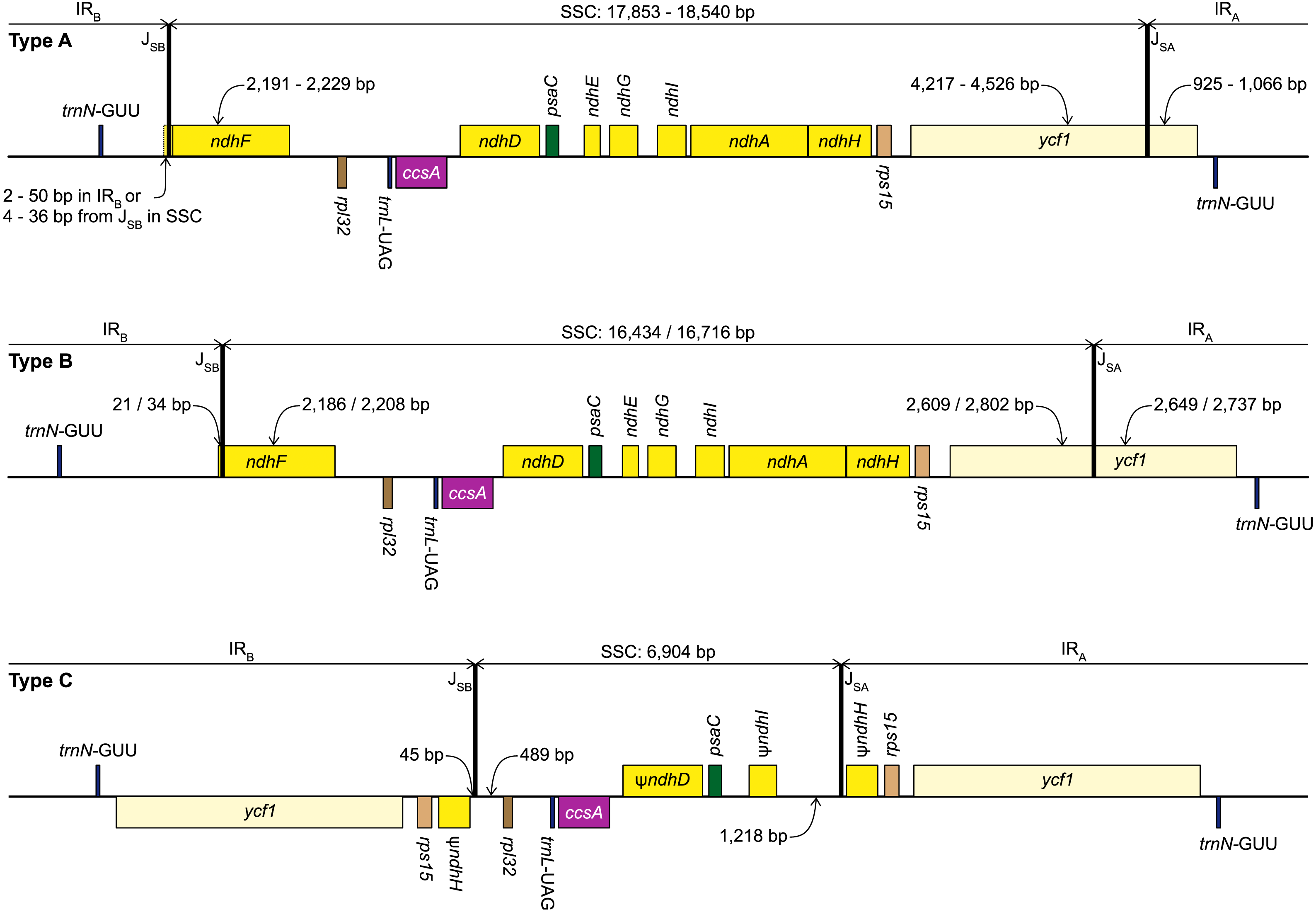{kind=link}
Protein coding genes were categorised as intact, putatively pseudogenised, or lost following Joyce et al. (2019). Briefly, genes were annotated as putative pseudogenes if they contained internal stop codons, or the stop codon was missing. Genes were considered lost if less than 30% of the gene was present compared with the other samples. Plastome maps were drawn in OGDRAW v1.3.1 (Greiner et al., 2011).
Results
Illumina pair-end sequencing for the samples in this study produced 19,015,437 - 24,547,050 raw paired-end reads. All assembly strategies produced consistent assemblies for P. maritimum. Fast-Plast with 20M reads and NOVOPlasty assemblies for Acis autumnalis var. oporantha were congruent. Variation in strategies for assembly for all other plastomes caused differences in overall length (Table S4), likely due to differing assembly of repeat rich regions. Notwithstanding, the structural parts of the plastome (LSC-IR-SSC) and expected genes could be identified in all output files, therefore final assemblies were constructed by hand. Sanger sequencing confirmed that the junctions in all five plastomes and the ndh genes in S. truncata were correctly assembled. Average coverage of the final assemblies ranged from 786 × to 1, 379 × (Table S4). Raw sequence data are available in SRA (BioProject: PRJNA730513); assembled plastomes are available on GenBank (MN539611–MN539615). Amaryllidaceae plastomes (Fig. 1) have a quadripartite structure, range from 153,129 to 160,123 bp in length, and contain 70–86 protein coding genes, 38 tRNAs and 8 rRNAs (Table 2).
| Amaryllidaceae | Asparagaceae | Asphodelaceae | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amaryllidoideae | Allioideae | Agapanthoideae | |||||||||||||||
| Acis autumnalisvar.oporantha | Lapiedra martinezii | Nerine sarniensis | Pancratium maritimum | Strumaria truncata | Clivia miniata | Hippeastrum rutilum | Leucojum aestivum | Lycoris radiata | Lycoris squamigera | Narcissus poeticus | Allium altaicum | Allium fistulosum | Allium praemixtum | Agapanthus coddii | Hesperoyucca whipplei | Hemerocallis fulva | |
| GenBank accession number | MN539611 | MN539612 | MN539613 | MN539614 | MN539615 | MN857162 | MT133568 | MH422130 | MN158120 | MH118290 | MH706763 | MH159130 | MH926357 | MK411817 | KX790363 | KX931459 | MG914655 |
| Total plastome length (bp) | 157,839 | 159,022 | 158,312 | 160,123 | 157,566 | 158,114 | 158,357 | 157,241 | 158,335 | 158,459 | 166,099 | 153,129 | 153,164 | 153,226 | 157,055 | 157,832 | 155,855 |
| LSC length (bp) | 85,792 | 86,756 | 86,271 | 86,393 | 85,648 | 86,204 | 86,451 | 85,657 | 86,613 | 86,431 | 86,445 | 82,196 | 82,237 | 82,162 | 85,204 | 86,170 | 84,607 |
| SSC length (bp) | 18,367 | 18,540 | 18,455 | 16,716 | 6,904 | 18,334 | 18,272 | 18,180 | 18,262 | 18,500 | 16,434 | 17,913 | 17,907 | 18,042 | 18,113 | 18,228 | 18,508 |
| IR length (bp) | 26,840 | 26,863 | 26,793 | 28,507 | 32,507 | 26,788 | 26,817 | 26,702 | 26,730 | 26,764 | 28,610 | 26,510 | 26,510 | 26,511 | 26,869 | 26,717 | 26,370 |
| Overall G/C content | 37.7 | 37.8 | 37.8 | 37.8 | 37.8 | 38 | 37.9 | 37.9 | 37.8 | 37.8 | 37.8 | 36.8 | 36.8 | 36.8 | 37.5 | 37.8 | 37.4 |
| Number of tandem repeats | 562 | 605 | 601 | 556 | 591 | 568 | 560 | 567 | 579 | 572 | 543 | 553 | 564 | 561 | 570 | 576 | 666 |
| Sum length of tandem repeats (bp) | 7,017 | 7,939 | 7,922 | 6,875 | 7,591 | 7,024 | 7,021 | 7,178 | 7,140 | 7,029 | 6,838 | 7,066 | 7,267 | 7,254 | 6,975 | 7,019 | 8,790 |
| Percentage length of tandem repeats | 4.45 | 4.99 | 5.00 | 4.29 | 4.82 | 4.44 | 4.43 | 4.56 | 4.51 | 4.44 | 4.12 | 4.61 | 4.74 | 4.73 | 4.44 | 4.45 | 5.64 |
| N of Intact protein coding genes (unique) | 86 (79) | 86 (79) | 84 (77) | 85 (78) | 79 (70) | 86 (79) | 86 (79) | 86 (79) | 85* (79) | 86 (79) | 85 (78) | 85 (78) | 85 (78) | 85 (78) | 86 (79) | 86 (79) | 86 (79) |
| Pseudo protein coding genes | 0 | 0 | 2 | 1 | 5 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 |
| Lost protein coding genes | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| N of Intact tRNA (unique) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) | 38 (30) |
| N of Intact rRNA (unique) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) |
Notes:
We identified three different JSA structures (Fig. 1) in our samples. Type A was the most frequent junction type, found in 14 samples, and most plausibly the ancestral IR junction in Amaryllidaceae, as it is shared with both outgroups. The other two JSA structures show two independent IR expansion events. Type B, present in Na. poeticus and P. maritimum, is a shared IR expansion event, while Type C in S. truncata represents a different IR expansion. The 5′ portion of ycf1 within the IRA ranged from 925–1,066 bp for Type A (Fig. 2). The Type B plastome had 2,649 or 2,737 bp of ycf1 included in IRA. The IR expansion in Type C has a different gene arrangement (Fig. 2): the entire ycf1, rps15, and pseudogenised ndhH were included in the IR. The IRs and the SSC ranged from 17,853 to 18,540 bp and 26,370 to 26,869 bp, respectively, in Type A plastomes. The IRs in the Type B plastomes were longer, at 28,507 or 28,610 bp, with a shorter SSC, of 16,434 or 16,716 bp. The expanded IRs in S. truncata (Type C) were the longest at 32,507 bp, while the 6,904 bp long SSC was the shortest. The arrangement of the junction between SSC and IRB also showed variation in Type A and Type B plastomes, however this was due to the length variation of the 3′ end of ndhF rather than IR expansion/contraction.
Repeat Finder identified only one short inverted repeat, with our search parameters, in P. maritimum (Table 3). A similar repeat is present in the Na. poeticus plastome, however this has more mismatches than the 10% threshold we set for the search. Both Type B plastomes contain the same, short, forward repeat, but in Na. poeticus this repeat is only partially in our target region. No short repeats were identified in the S. truncata or Ne. sarniensis plastomes. Figure 3 summarises the putative mechanisms for the development of the Type B and C plastomes.
| Species | Repeat sequence | Direction | Repeat length | Position | Mismatched bases |
|---|---|---|---|---|---|
| P. maritimum | TCATTATTAGGTTTATA - TATAAACCCAATAATGA | inverted | 17 bp | 131,409–131,425 133,062–133,078 | 1 |
| P. maritimum | TTCATTTTTCTCTTCTTT - TTCATTTTCCTCTTCTTT | forward | 18 bp | 132,022–132,039 133,345–13,336 | 1 |
| Na. poeticus | TCATTATTAGGTTTATA - TATAAACCCAATAATTA | inverted | 17 bp | 131,379–131,395 133,023–133,039 | 2 |
| Na. poeticus | TTCATTTTTCTCTTCTTT - TTCATTTTCCTCTTCTTT | forward | 18 bp | 131,983–132,000 133,306–133,323 | 1 |
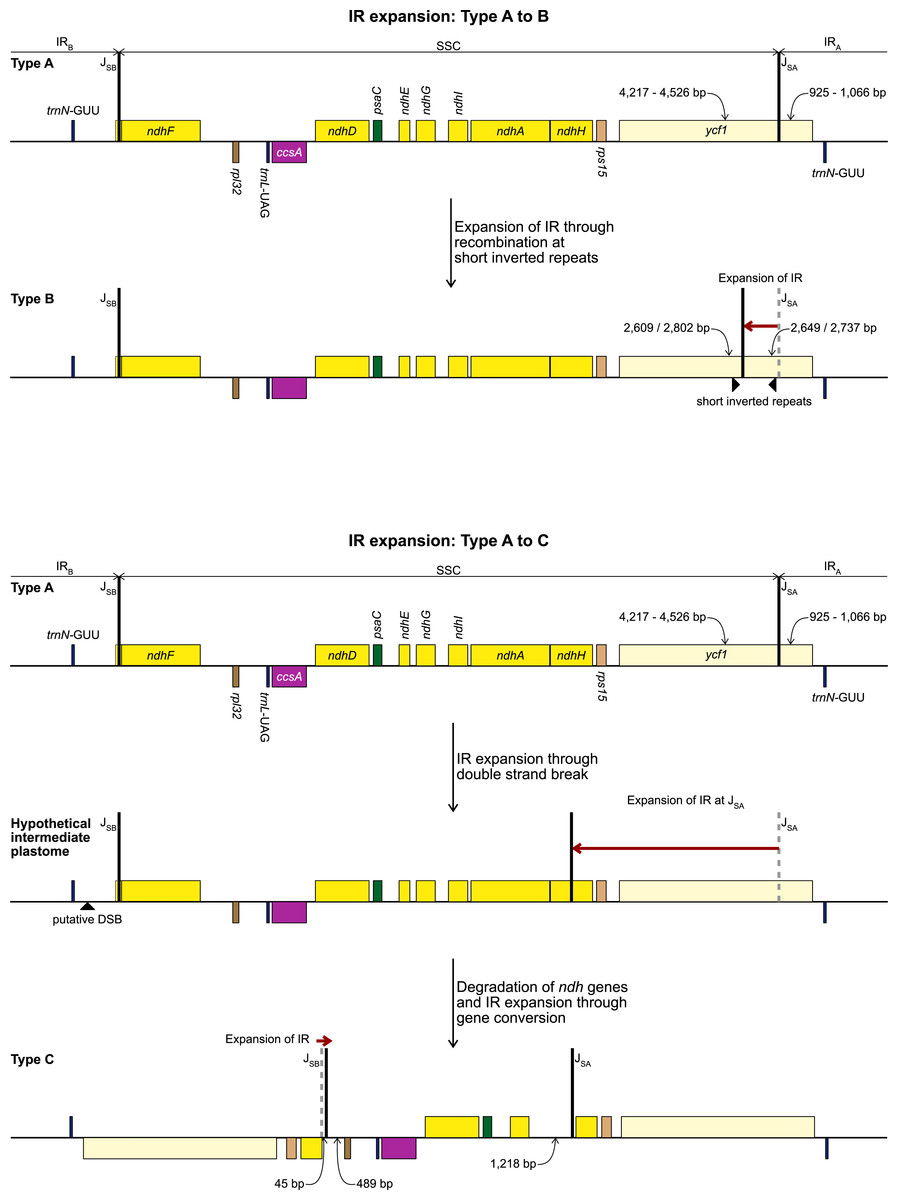
Figure 3: Development of Type B (top) and Type C (bottom) plastome from the ancestral Type A plastome.
Steps required to explain the structural changes and the putative mechanisms are detailed in the figure. Gene order, direction of transcription, and colour code of each gene correspond to Fig. 1.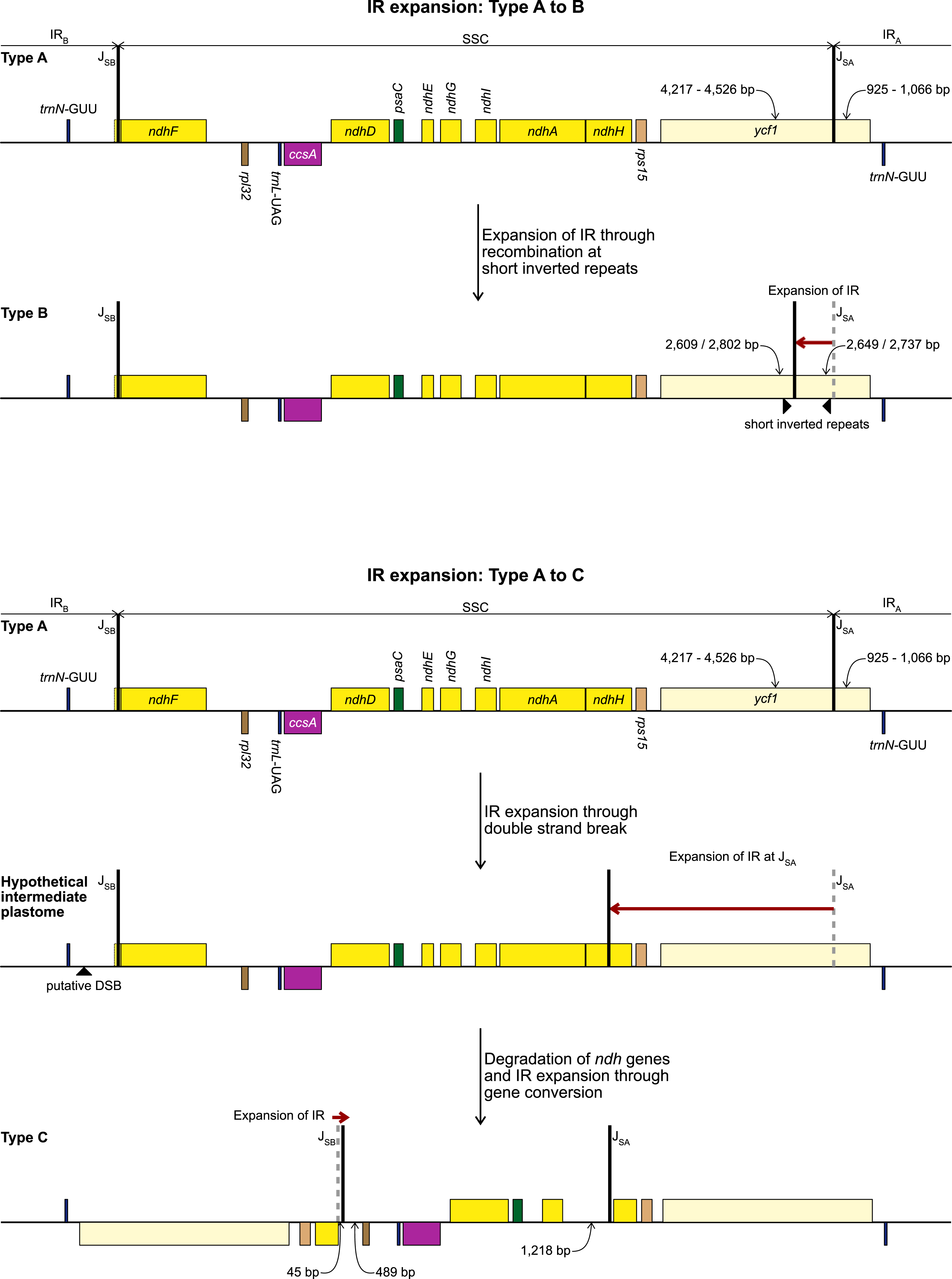{kind=link}
The alignment of the regions between the two copies of trnN encompassing the SSC from Ne. sarniensis and S. truncata showed that the 45 bp present in the S. truncata IR and the following single-copy sequence towards rpl32 is homologous with the ndhF-rpl32 spacer in Ne. sarniensis (Fig. S1). These 45 bp represent a second short independent IR expansion in S. truncata (Fig. 3).
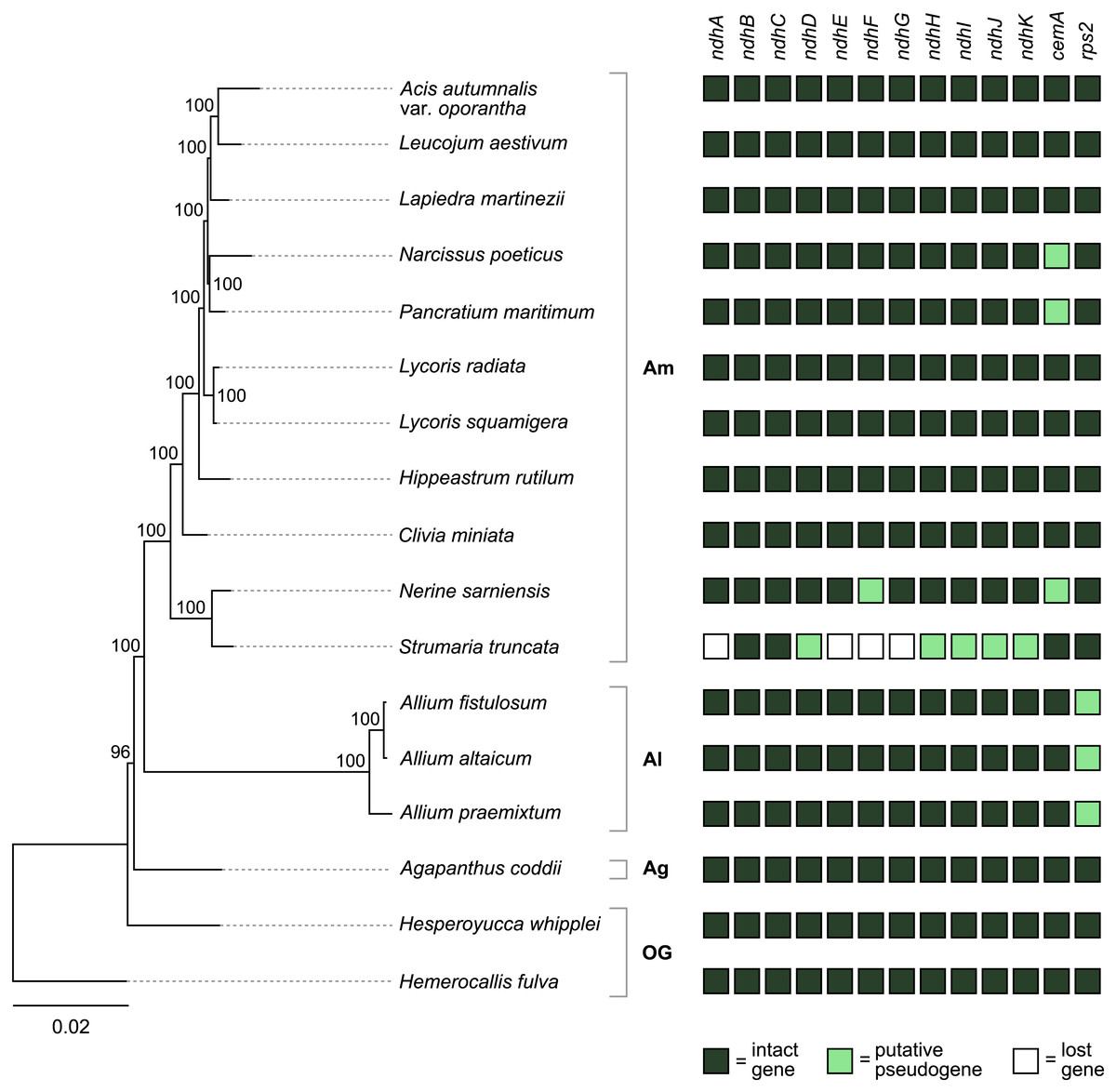
Figure 4: Pattern of gene losses and pseudogenization in Amaryllidaceae and the two outgroup samples plotted against the RAxML tree based on 67 coding sequences.
Bootstrap support values are shown at nodes. Plastomes assembled in this study are highlighted in bold. Amaryllidaceae subfamilies are indicated on the right: Am, Amaryllidoideae; Al, Alliodideae; Ag, Agapanthodieae; OG, Outgroups.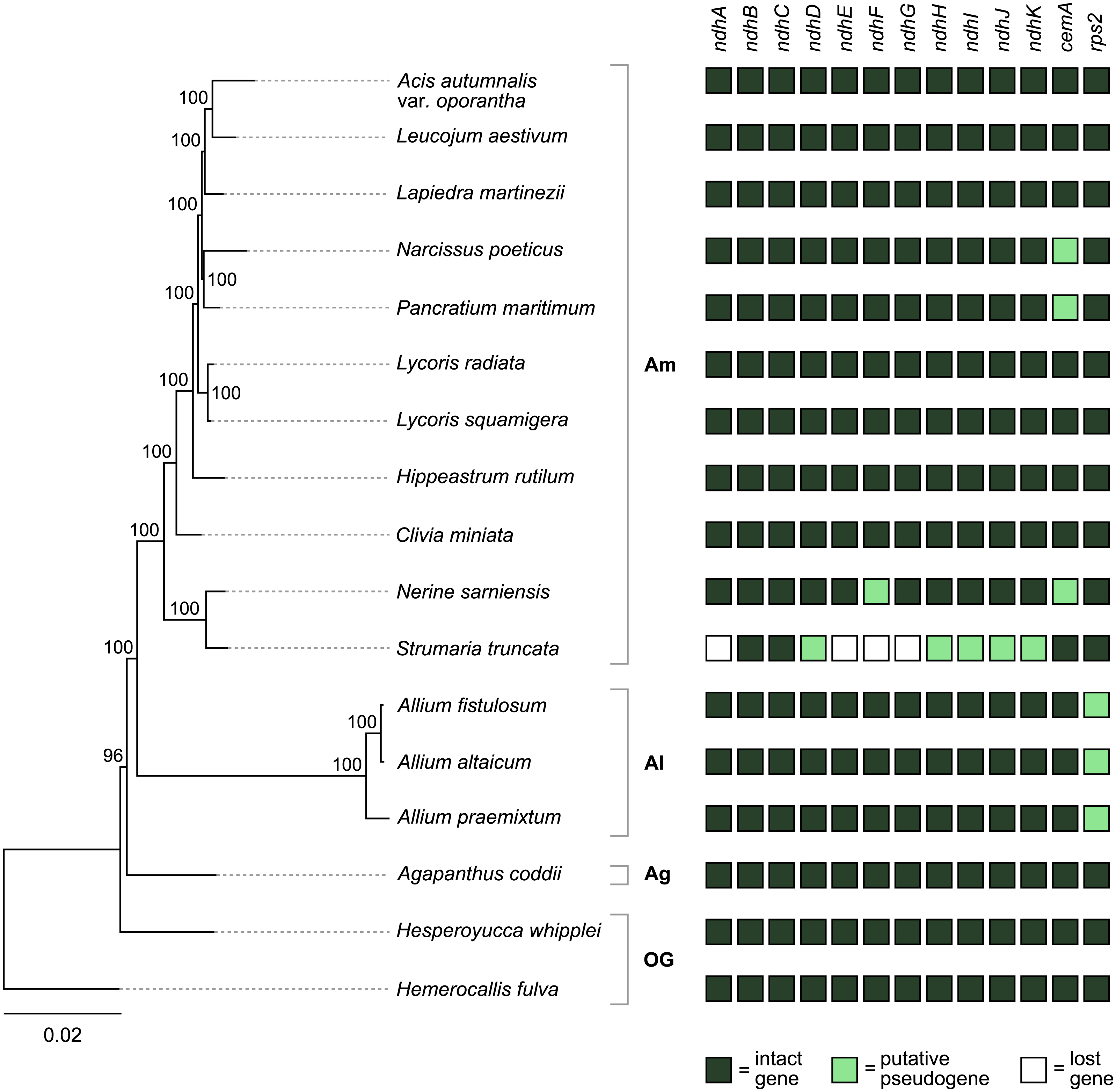{kind=link}
The S. truncata plastome (Type C) showed a degradation of the ndh-suite (Fig. 4). Only two ndh-genes remained intact, ndhB and ndhC. The ndhA, ndhE, ndhF, and ndhG genes were lost, while ndhD, ndhH, ndhI, ndhJ, ndhK were pseudogenised. The combined length of the two putative exons of ndhA is 21% of the length of ndhA in other Amaryllidaceae samples. The sequence regions of the other genes classified as lost (ndhE, ndhF, ndhG) were entirely absent. The ndhH gene is 1,182 bp long in Amaryllidaceae. In S. truncata, this gene is included in the IR as two putative pseudogenes, each copy is only 592 bp long (50%) from the start codon, and the stop codons are missing. The ndhJ gene contained an internal stop codon due to transversion mutation (G to T at 49,624 bp), all other putative pseudogenes had frameshift mutations. Furthermore, ndhF was classified as a putative pseudogene in Ne. sarniensis due to a missing stop codon. The ndhC gene in Leucojum aestivum is missing 47 bp, including the start codon, at the 5′ end of the gene preceded by 2 N’s, potentially indicating scaffolded contigs due to missing data. We did not categorise this gene as a putative pseudogene due to this potential missing data, however we did exclude the gene from the phylogenetic analysis. The cemA gene contains frameshift mutations in Na. poeticus, Ne. sarniensis, and P. maritium due to a homopolymeric A repeat at the 5′ end of the gene. In all Allium samples rps2 is a pseudogene.
The number of tandem repeats and their length as percentage of the total plastome ranged from 543 to 605 and 4.1% to 5.6%, respectively, in Amaryllidaceae (Table 2). Tandem repeat numbers in Type A plastomes ranged from 560 to 601 (4.4–5.0%) (Table 2). Type B plastomes had 543 (4.1%), and 556 (4.3%) tandem repeats in Na. poeticus and P. maritimum, respectively, and 591 (4.8%) in S. truncata (Type C). Moreover, the highest number of repeats, 666, was found in Hemerocallis fulva, which shows no IR expansion. These changes indicate that an increased number of tandem repeats did not contribute to the IR expansions in Type B and C plastomes.
The phylogenetic analysis based on 67 protein coding genes recovered a fully resolved phylogenetic tree with maximum (100%) bootstrap support for all branches except for the one leading to the ingroup (96%, Fig. 4). Within Amaryllidaceae, Agapanthoideae is sister to Allioideae and Amaryllidoideae. Within Amaryllidoideae, Ne. sarniensis is sister to S. truncata and in turn they are sister to the remaining Amaryllidoideae species. Sequence alignment of the 67 protein coding genes used in the phylogenetic analysis and the resulting newick tree is available at: https://github.com/kalmankonyves/Amaryllidaceae_plastomes-Strumaria.
Discussion
The newly sequenced and assembled plastomes of five genera from Amaryllidoideae exhibited three plastome arrangement types based on the portion of ycf1 within the inverted repeats. Previously published plastomes such as Allium (Huo et al., 2019) and Narcissus (Könyves et al., 2018) fall into our Type A and B respectively, however the Type C plastome, with the largest IR expansion, represents a novel rearrangement in Amaryllidaceae that has not been reported previously.
In typical angiosperm plastomes ∼1,000 bp of ycf1 is included in the IR (Sun et al., 2017). This is similar to the Type A plastome we recognised in Amaryllidaceae. We identify the Type A plastome as the ancestral state in the family, as it is shared with both outgroups (Fig. 1). The IR expansions that gave rise to the Type B and Type C plastomes represent independent events. In Na. poeticus and P. maritimum (Type B) the IR has expanded to include a larger portion of ycf1 (2,649/2,737 bp), while in S. truncata (Type C) the whole of ycf1 along with rps15 and a pseudogenised ndhH are contained within the IR. The expansion or contraction of the inverted repeats have been shown to occur in multiple land plant lineages (Wicke et al., 2011; Jansen & Ruhlman, 2012; Zhu et al., 2016) and can often be specific to a few genera within a family (Guisinger et al., 2011; Dugas et al., 2015; Tian et al., 2018; Thode & Lohmann, 2019). In Asparagales IR expansions have been reported in multiple genera in Orchidaceae, incorporating genes from the SSC up to and including ccsA (Kim et al., 2015; Kim et al., 2020), and in Eustrephus latifolius, Asparagaceae (Kim, Kim & Kim, 2016), where the IR expanded to include ycf1.
Rearrangements in the plastomes have been associated with an increased number of repeats (Guisinger et al., 2011; Sinn et al., 2018). There was no correlation between the number of tandem repeats and IR expansion in Amaryllidaceae; the tandem repeat numbers and their length as a percentage of the total plastome (543–605; 4.1%–5.6%) are similar to those reported by Sinn et al. (2018) for non-rearranged plastomes (420–732; 3.3%–6.0%). However, we identified 17 bp inverted repeats in the vicinity of the IR junctions in both P. maritimum and Na. poeticus. We propose that the IR expansion in these species might have happened through recombination of similar short inverted repeats in the common ancestor of these genera (Fig. 3). The literature offers no consensus on how long short inverted repeats are. The earliest report by Palmer et al. (1985) for Chlamydomonas reinhardtii, identified 100–300 bp repeats in the vicinity of the IR junctions, however Aldrich et al. (1988) found 7 bp inverted repeats in Petunia and hypothesised that recombination at these repeats led to the IR expansion. We based our search for short inverted repeats on Staub & Maliga (1994) who showed experimental evidence of recombination at 16 bp imperfect repeats in Nicotiana tabacum resulting in extrachromosomal elements.
No short repeats were identified in the S. truncata or Ne. sarniensis plastomes. Therefore, we hypothesise that the IR expansion in S. truncata could be a result of a double-strand break repair (Fig. 3) as described by Goulding et al. (1996) in Nicotiana or homologous recombination between different plastome units could also have produced the expanded IR. Choi, Jansen & Ruhlman (2019) proposed that short non-allelic repeats mediated recombination in Medicago resulting in the reestablishment of the inverted repeats. As we did not find any suitable repeats, the recombination instead could have happened at homologous genes as shown by Ruhlman et al. (2017) in Monsonia. It is also possible that any site of recombination could have been lost during the degradation of the ndh genes. We identified a 45 bp region downstream of the annotated ndhH pseudogene to have originated in the ndhF-rpl32 spacer. This indicates that the IR-SSC organisation of S. truncata is the result of two independent IR expansion events (Fig. 3). Most likely, first a double-strand break originating in IRB got repaired against the complementary strand of IRA with the copy-repair progressing beyond the IRA-SSC junction incorporating ycf1, rps15, and 592 bp of ndhH into the IR. A second small IR expansion originating in IRB further expanded the IR incorporating 45 bp of the region upstream of rpl32 through the gene conversion mechanisms of Goulding et al. (1996). Although Goulding et al. (1996) called the mechanism responsible for short IR expansion ‘gene conversion’, their description: “Branch migration of a Holliday junction across an IR/LSC junction results in the formation of heteroduplex before this process stalls. Resolution of heteroduplex may then proceed by sequence correction against either strand…” does not preclude this model from applying to expansions at SC/IR junctions with non-coding sequences. Furthermore, ‘gene conversion’ at non-coding sequences removes the constraint of maintaining functional genes.
In S. truncata nine out of the 11 ndh genes have been lost or pseudogenised (Fig. 4). Loss or pseudogenisation of ndh genes has been reported in Asparagales both in mycoheterotrophic and autotrophic orchids (Barrett et al., 2014; Kim et al., 2015; Kim et al., 2020; Roma et al., 2018), and in Allium paradoxum (2019). We found a further putative ndh pseudogene, ndhF in Ne. sarniensis, which is missing the stop codon. It is possible that this gene is functional and C-to-U RNA editing creates a stop codon at transcription. The pseudogenes in S. truncata, on the other hand, are the result of frameshift mutations which are not known to be edited in plastomes (Haberle et al., 2008). The ndh genes encode the NADH dehydrogenase-like complex (NDH-1) which regulate photosynthetic electron transport (Peltier, Aro & Shikanai, 2016; Shikanai, 2016). NDH-1 helps adapt photosynthesis under photooxidative stress conditions (Martín & Sabater, 2010) and in environments with fluctuating light intensity (Yamori & Shikanai, 2016). The loss of the ndh complex has been reported in plants growing either in high or low light level environments, for example hot desert (Sanderson et al., 2015), submersed (Peredo, King & Les, 2013), or understorey habitats (Graham, Lam & Merckx, 2017; Omelchenko et al., 2020). Furthermore, under optimal growth conditions ndh genes appear dispensable (Burrows et al., 1998; Endo et al., 1999; Rumeau, Peltier & Cournac, 2007), and their function could also be assumed by an alternate nuclear encoded system (Wicke et al., 2011) e.g., the AA-sensitive CEF pathway (Sanderson et al., 2015). In the presence of an alternate system, the degradation of the ndh genes can have little consequence on plant fitness and mutations can freely accumulate leading to pseudogenisation and loss. Alternatively, ndh genes can be transferred to the nucleus and only lost from the plastome, as Roma et al. (2018) showed in Ophrys. The data we present in this paper, shallow sequencing of whole genomes (e.g., ∼6.3 Gb of raw sequence data represent ∼0.3–0.5× coverage of the nuclear genome of Ne. sarniensis: 12.6/19.4 Gb; Leitch et al. (2019)), allows us to simply hypothesise the fate of pseudogenised and lost genes. The loss of ndh genes from the S. truncata plastome compared with Ne. sarniensis could be the result of ecological adaptation, as the two species occur in different habitats: S. truncata inhabits the semi-arid Succulent Karoo, while Ne. sarniensis occurs in the Fynbos, characterised by a wetter, Mediterranean-type climate (Duncan, Jeppe & Voigt, 2020). It is also possible that the loss of the ndh genes is a result of a transfer to the nucleus, or the presence of an alternate system. Further sampling of Strumaria and Nerine species and investigating the nuclear genome and transcriptome will be necessary to identify potential routes of gene loss from the plastome.
Kim et al. (2015) and Kim et al. (2020) showed a correlation between the presence of ndhF and the organisation of the IR-SSC junctions in Orchidaceae, and proposed that the loss of ndhF leads to destabilisation of the IR-SSC junctions. This holds true to Najas flexilis, mentioned by Kim et al. (2015), and could be supported by our results. However, there is also published evidence that IR expansion can happen without the loss of ndh genes: plastomes in Thymelaeaceae have all SSC genes, apart from ndhF and rpl32, incorporated in the IR in the presence of a full set of 11 ndh genes (Könyves et al., 2019b; Lee et al., 2020; Liang, Xie & Yan, 2020), and in Asarum the IR expanded to encompass the whole of the SSC without the loss of ndh genes (Sinn et al., 2018).
The IR expansion in S. truncata is plausibly the result of two independent events, therefore the order in which gene losses and expansions happened are difficult to unravel. It is perhaps more parsimonious to suggest that the degradation of the ndh genes or at least the loss of ndhF preceded the second IR expansion in S. truncata, otherwise the whole of ndhF had to be incorporated into the IR and subsequently lost from both IRA and IRB. Further sampling in Amaryllidoideae could help establish the sequence of gene loss and IR expansion and any correlation between these events.
There were further pseudogenised genes found in Amaryllidaceae: the chloroplast envelope membrane protein encoding cemA is pseudogenised in Ne. sarniensis, Na. poeticus, and P. maritimum due to a homopolymeric A repeat adjacent to the potential initiation codon. A similar pattern of pseudogenisation has been shown in Lilium spp. (Do & Kim, 2019) and Cocos nucifera (Huang, Matzke & Matzke, 2013). The cemA gene is not essential for photosynthesis, however in high light conditions cemA-lacking mutants of Chlamydomonas reinhardtii showed decreased photosynthetic efficacy (Rolland et al., 1997). The rps2 gene is functional in Amaryllidoideae and Agapanthus coddii but has pseudogenised due to internal stop codons in Allium, which has been reported previously by Filyushin, Beletsky & Kochieva (2019), Omelchenko et al. (2020) and Xie et al. (2019).
The phylogenetic relationship between the subfamilies recovered in this study: Agapanthoideae sister to an Allioideae-Amaryllidoideae clade is congruent with previously published studies based on plastid DNA data (Fay et al., 2000; Givnish et al., 2005; Givnish et al., 2018; Pires et al., 2006; Seberg et al., 2012). Moreover, while our sampling represents only a small fraction of the diversity within Amaryllidoideae, the relationship within the subfamily is also broadly congruent with previous studies (Meerow et al., 2006); Rønsted et al., 2012). The long branch leading to Allium in our phylogenetic tree is also present in studies using fewer (one to four) plastid genes (Givnish et al., 2005; Seberg et al., 2012; Chen et al., 2013); the reason behind this is beyond the scope of our paper.
Conclusions
We are still very much in the discovery stage when it comes to patterns of gene loss and duplication in the plastome. Genes linked to a range of functions are implicated in plastome change however, not surprisingly, most of the genes that vary are linked to photosynthesis at one level or another. The loss of so many genes is Strumaria truncata is notable and worthy of further phylogenetic investigation through deeper taxon sampling. At this stage we are not in a position to propose driving mechanisms for the change, or whether those observed are to some extent evolutionarily neutral to the growing conditions the plants experience. While the family is characterised by bulbous geophytes, the habitats those plants occupy vary enormously from arid through mesic to seasonally inundated. Likewise, the light levels tolerated by different species vary from intense sun to quite deep shade. This paper offers a first look at the kinds of variation in plastomes that might be found across the family and indicates this will be a promising area for more detailed investigation.
Supplemental Information
Alignment of Strumaria truncata and Nerine sarniensis plastomes in Geneious Prime 2020.0.5, showing: (A)the extracted regions between trnN and trnN in the IRs, including the complete SSC; (B) JSB and (C) J[sub]
Red boxes highlight the alignment of the 45 bp sequence region downstream of the ndhH pseudogene present in the S. truncata IRs. Green bars under ‘Consensus’ indicate 100% pairwise sequence identity, greeny-brown bars > 30% and red bars < 30% pairwise sequence identity. Lack of any bars indicate SNPs and indels in panels B and C. Green arrows indicate gene annotations, yellow arrows show protein coding reading frames, purple arrows show tRNAs, and orange arrows are IR annotations.
Details of the PCR primers used to amplify and sequence junctions of the inverted repeats and the ndh gene sequences in the Strumaria truncata plastome assembly
Primers for this study were designed with the Primer3 plugin in Geneious v11.1.5. PCR primers are highlighted in bold, all other primers used as internal sequencing primers.