
X-ray diffraction and theoretical study of the transition 2H-3R polytypes in Nb1+xSe2 (0 < x < 0.1)
- Published
- Accepted
- Received
- Academic Editor
- Jordi Cirera
- Subject Areas
- Crystallography, Theoretical and Computational Chemistry, Transition Metal Chemistry
- Keywords
- Niobium diselenide, Polytype, Non- stoichiometry, X-ray diffraction, CASTEP
- Copyright
- © 2021 Sidoumou et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Inorganic Chemistry) and either DOI or URL of the article must be cited.
- Cite this article
- 2021. X-ray diffraction and theoretical study of the transition 2H-3R polytypes in Nb1+xSe2 (0 < x < 0.1) PeerJ Inorganic Chemistry 3:e2 https://doi.org/10.7717/peerj-ichem.2
Abstract
Single crystals of 2H and 3R niobium diselenide were grown by a chemical transport reaction. The current determinations by single crystals X-ray diffraction reveal a non-stoichiometric composition. The structures are built from Se—Nb—Se slabs with Nb in trigonal prismatic coordination whereas the extra or additional Nb atoms are located in the octahedral holes between the slabs giving rise to the formula 2H and 3R-Nb1+xSe2 with 0.07 < x < 0.118. In particular, vacancy and Nb-Nb interactions may play an important role on the non-stoichiometry and the stacking mode in NbSe2. By increasing the number of additional Nb atoms in the pure 2H-NbSe2, a transition 2H to 3R polytype should occur in order to minimize the Nblayer—Nbextra—Nblayer repulsions between these adjacent slabs. The theoretical study shows that both 2H and 3R-Nb1+xSe2 are thermodynamically stable in the range 0 < x < 0.1.
Introduction
NbSe2 belongs to the transition metal dichalcogenides TMDC’s with chemical formula TX2 (T = Nb, Ta; X = S, Se), which have recently renewed interest because of their quasi 2D nature similar to graphene making them very promising in novel electronic devices applications (Vogel & Robinson, 2015). The system NbSe2 has been the subject of many investigations since it exhibits incommensurate charge density waves (CDW) and superconductivity phenomenon above 4K (Wilson, Disalvo & Mahajan, 1975).
The structure consists in hexagonally arranged Se-Nb-Se sandwiches with metal atoms (Nb) that are located between two layers of chalcogen atoms (Se) in a trigonal prismatic coordination. The bonding within each sandwich is covalent while the bonding among sandwiches themselves is a weak Van-der Waals type. Although the stacking along the c axis gives rise to several polytypes usually described as 1T–NbSe2, 2H- NbSe2, 3R–NbSe2 and 4H- NbSe2 (Kalikhman & Umanskii, 1973; Brown & Beerntsen, 1965), it seems that only the 2H and 3R are frequently obtained in practice.
Until date, there are many reports on polymorphism in the TMDC’s by tuning synthesis temperature such as the high temperature monoclinic MoTe2 (Brown, 1966), the layered telluride Mo1-x NbxTe2 (Ikeura et al., 2015), polytypism in TiS2 which was first observed by Tronc & Huber (1973) and Legendre et al. (1983), the highly polymorphic TaSe2 (Brown & Beerntsen, 1965; Huisman, Kadijk & Jellinek, 1970) and the polymorph TaSe2-xTex (Luo et al., 2015).
This structural anisotropy enables the intercalation of species to form stoichiometric and non-stoichiometric compounds with different guests. A number of authors (Huisman, Kadijk & Jellinek, 1970) and references therein) reported the existence of the Nb self-intercalation (SI) in NbSe2 phases where the additional Nb metal atoms lie in octahedral holes between NbSe2 layers. Most of the structures were characterized by X-ray powder diffraction method, and very few investigations using single crystal X-ray diffraction technique were performed.
The Nb-Se phase diagram has been established on the basis of the homogeneity range of several polytypes which mostly depend on the method and the synthesis conditions (Predel, 1997). However, and according to the recent findings (Ivanova et al., 2019), the diagram may contain some ambiguous data and should be revisited
The selenide 2H-NbSe2 revisited by single crystals (Meerschaut & Deudon, 2001) and the selenide 3R-NbSe2 investigated on the basis of the precession photograph (Huisman, Kadijk & Jellinek, 1970) have been both found stoichiometric; while the sulfur polytype 3R-Nb1+xS2 (x = 0.06 and 0.09) (Meerschaut & Deudon, 2001; Powell & Jacobson, 1981) is non-stoichiometric.
According to Fisher & Sienko (1980), these compounds are non-stoichiometric. Indeed, in the 2H-NbSe2, the niobium atoms are stacked directly one above the other along the c axis, which is not the case for 3R-NbSe2. In the case of an excessive intercalate niobium a transition from 2H to 3R polytype should then occur in order to minimize the Nblayer—Nbextra—Nbextra repulsions between these adjacent slabs. This transition was explained by an NbS2-layers rotation mechanism involving stacking faults probabilities between 15–18% (Katzke, 2002; Leroux et al., 2018).
In this work, three polytypes in the Nb-Se system: 2H-Nb1.031Se2, 3R-Nb1.071Se2 and 3R-Nb1.085Se2 were synthesized by CVT and investigated by single crystal X-ray diffraction.
In particular, the role of vacancy and Nb-Nb interactions on the non-stoichiometry and stacking mode in NbSe2 has been examined.
Both 2H and 3R polytypes may co-exist in the range 0<x < 0.07 and above this limit where only the form 3R predominates as a transition 2H-3R will take place.
Furthermore, by using DFT with CASTEP code, a comparative study involving thermodynamic polytype stability of 2H and 3R-Nb1+xSe2 (x = 0, 0.1) has been attempted in order to support the X-ray diffraction conclusions.
Materials & Methods
Crystal growth and chemical analysis
Single crystals of 2H and 3R-Nb1+xSe2 were obtained by chemical vapor transport (CVT) method during our attempts to prepare ternary HgNbSe2 and SnNbSe2. A stoichiometric amount of pure elements with an excess of Se were mixed and sealed in an evacuated quartz tube (length ∼18 cm) using iodine (<5 mg/cm3) as a transport agent to favour crystallization. The mixtures (charge ∼1g) were placed into a zone tube furnace. The temperature was first increased slowly at 500 °C and held then for 48 h in order to avoid thermal runaway caused by the exothermic reaction between Nb and Se. After that the temperature is turned up between 760−1,050 °C for 15 days. Finally the furnace was allowed to cool slowly to room temperature. Crystals of appreciable sizes were grown in the cold end of the tube; 2H+3R crystals are obtained at ∼760 °C while the 3R crystals are obtained at ∼1,050 °C. The analysis results by X-ray energy dispersive spectroscopy (XEDS) with the TEM (EDX Oxford Instruments) on several different crystallites of each polytype sample gave an approximate atomic ratio of 1:2 for Nb and Se [for a selected 3R-crystal: Nb (at%) = 29,9 and Se (at%) = 70,1].
Single crystal structure determination
Single crystals data were recorded on a Kappa Apex II CCD X-ray diffractometer (Bruker, 2006) with graphite-monochromated MoKα (λ = 0.71071 Å) radiation. The reflection intensities were integrated with the SAINT (Bruker, 2006); SADABS was used for empirical absorption correction (Sheldrick, 2002, and Petříček, Dušék & Palatinus, 2006) was performed for the structure refinement.
The crystal structure of 3R-Nb1.071Se2 was obverse/reverse twinned while the 3R-Nb1.085Se2 was twinned by inversion; with a refined twin domain fraction of 0.611(5): 0.389(5) and 0.85(8): 0.15(8) respectively. The CIF files containing details of the structure refinements are available in the File S1.
In the final cycles of refinement, all the atoms in the different structures were refined anisotropically.
Details concerning the structure refinement and final results are presented in Table 1 while atomic coordinates, anisotropic displacement parameters and selected bond distances are listed in Tables S1–S3.
| Nb1+xSe2Polytype | 2H-Nb1.031Se2 | 3R-Nb1.071Se2 | 3R-Nb1.085Se2 |
|---|---|---|---|
| Molar mass (g.mol−1) | 253.7 | 257.3 | 258.1 |
| Crystal size (mm3) | 0.36 × 0.16 × 0.05 | 0.33 × 0.31 × 0.015 | 0.20 × 0.19 × 0.03 |
| Space group, Z | P63/mmc, 2 | R3m, 3 | R3m, 3 |
| Unit cell dimensions (Å) |
a = 3.4475(3) c = 12.5702(11) |
a = 3.4512(4) c = 18.827(3) |
a = 3.4670(4) c = 18.866(2) |
| c/a | 3.646 | 5.455 | 5.441 |
| (c/n)/a | 1.823 | 1.818 | 1.836 |
| Volume (Å3) | 129.38(2) | 194.21(4) | 196.39(4) |
| Calculated density (g cm−3) | 6.5101 | 6.5987 | 6.5442 |
| Absorption coefficient (mm−1) | 32.515 | 32.658 | 32.33 |
| Angular range θ (∘) | 3.24–27.48 | 6.50–39.95 | 3.24–29.86 |
| Index ranges | −3<h<4; −4<k<4 −16<l<16 |
−6<h<6; −6<k<4 −32<l<33 |
−4<h<4; −4<k<4 −25<l<25 |
| Total recorded reflections | 1568 | 1707 | 947 |
| Independent reflections, Rint | 80, 0.0505 | 547, 0.0498 | 188, 0.054 |
| Reflections with I >3σ(I) | 75 | 534 | 176 |
| Tmin/Tmax | 0.2910/0.7456 | 0.000/0.615 | 0.368/0.746 |
| Number parameters | 9 | 14 | 15 |
| R1, wR2 (all) | 0.0882/ 0.1033 | 0.0232/ 0.0271 | 0.057/ 0.084 |
| Flack parameter | 0.15(8) |
Theoretical calculations
The DFT calculations were performed using the plane-wave pseudopotential method implemented in the Cambridge Sequential Total Energy Package (CASTEP) code (Clark et al., 2005) of the Material Studio program from Accelrys (Materials Studio CASTEP Manual Accelrys 2010, 2010). The GGA-PBE functional (Hammer, Hansen & Nørskov, 1999) was used to model the exchange and correlation interactions. The Broyden–Fletcher–Goldfrab–Shanno (BFGS) method was used to carry out the geometrical optimization (Shanno, 1985). The plane-wave cut-off energy was adopted to be 500 eV and the Monkhorst–Pack scheme (Perdew, Burke & Ernzerhof, 1996), k-point grid sampling was set to 7 × 1 × 1 in the Brillouin zone. As far as self-consistent condition setting is concerned, the total energy was less than 3.10−2 eV atom, and the maximum displacement and maximum stress allowed was 10−3 Å and 5.10−2 GPa respectively. The valence electronic configurations considered for atomic pseudopotential calculation are Nb: 4s2 4p6 4d4 5s1 and Se: 4s2 4p4.
To investigate the relative stability of the Nb-SI in the 2H and 3R polytype with an excess of niobium (x = 0.1), 5 × 1 × 1 and 5 × 2 × 1 supercells respectively are adopted, corresponding to the ordered configurations Nb10Se20 (x = 0) and Nb11Se20 (x = 0.1) to simulate both pure and SI systems.
The formation enthalpy ΔE at T = 0 K is expressed as the difference in total energy of the supercell calculated by DFT and the chemical potentials E of Nb (cubic Fm-3m) and Se (trigonal P3121) calculated from their respective bulks in the same computation conditions. where x and y, respectively represent the Nb and Se atoms number in the supercell structure model.
The calculated free energy ΔG at typical synthesis temperatures of 760 and 1,050 °C only include configurational entropy due to intercalation, according to (Ivanova et al., 2019).
Results and Discussion
X-ray experimental crystal structure
The polytype 2H-Nb1.031Se2 is almost stoichiometric (Fig. 1A). Additional metal Nb2 atoms with a refined occupancy about 7.3% are located into the octahedral sites within the van der Waals VDW gap with the stacking sequence AcAδBcB (A,B refer to the selenide layers; c to the completely filled niobium layers and δ to the partially filled niobium layers).

Figure 1: Crystal structure of (A) 2H-Nb1.031Se2; (B) 3R-Nb1.085Se2 showing the prismatic packing of Nb1Se6 polyhedron (drawn in blue) and Nb2Se6 (drawn in purple).
The extra Nb2 atoms are located into the octahedral sites (Nb2Se6 drawn in purple) within the van der Waals gap.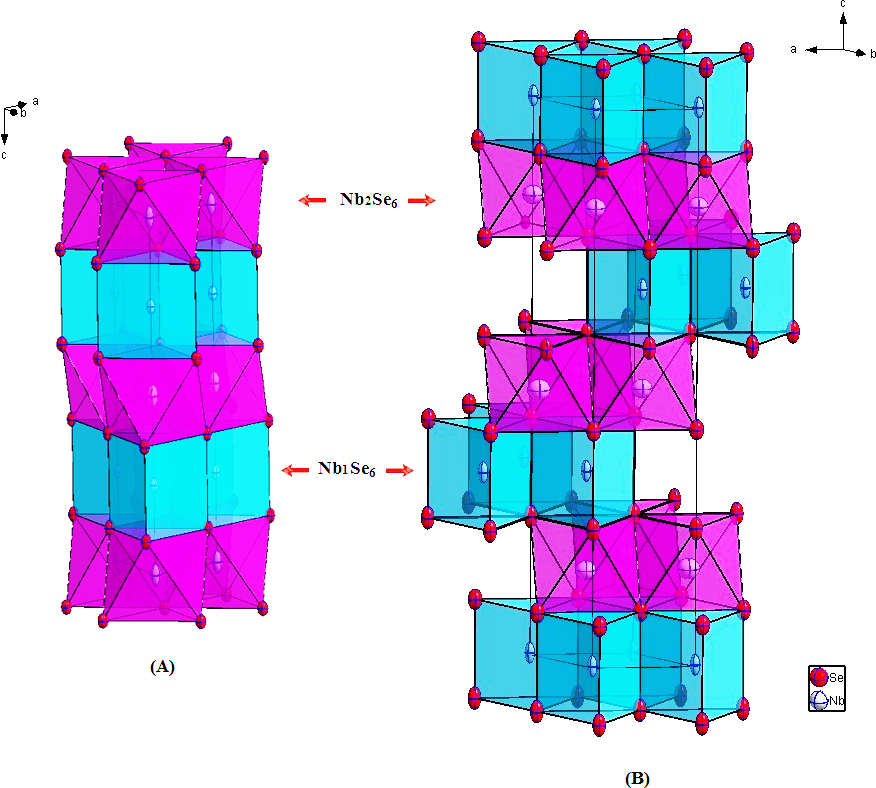{kind=link}
Polyhedrons around the Nb atoms are not distorted with six equivalent Nb-Se distances. The Nb1- 6Se = 2.597(2) Å around Nb1 (in a trigonal prismatic coordination) are somewhat larger than the Nb2-6Se = 2.477(2) Å, observed around Nb2.
A characteristic feature of this 2H- structure is the short Nb1-Nb2 = 3.1425(6) Å distance, suggesting the formation of strong metal–metal repulsions between the adjacent layers, but can also be analyzed in terms of electrostatic repulsion (Fig. 2A).
![Projection of the crystal structures of (A) 2H-Nb1.031Se2; (B) 3R-Nb1.085Se2 along the [100] direction, showing the Nblayer—Nbextra—Nblayer repulsions (red arrows).](https://dfzljdn9uc3pi.cloudfront.net/2021/ichem-2/1/fig-2-2x.jpg)
Figure 2: Projection of the crystal structures of (A) 2H-Nb1.031Se2; (B) 3R-Nb1.085Se2 along the [100] direction, showing the Nblayer—Nbextra—Nblayer repulsions (red arrows).
Thermal ellipsoids are drawn at 95% probability displacement level.{kind=link}
The relaxation probably occurs following a defect model by introducing vacancy (about 4%) in the Nb1 filled layers. The Nb1 atoms are then displaced forward the created vacancy to minimize the formation of Nb1-Nb2 pairs interactions (Tronc & Moret, 1981; Amzallag et al., 2007).
The composition obtained with the structure refinement is: 2H-(Nb0.96(2) Δ0.04) Nb0.073(13)Se2
(Δ denotes Nb vacancy in the fully layers) (ie) 2H-Nb1.031Se2.
The Nb1-Se distances in the 3R-Nb1.071Se2 polytype are very comparable [2.594(9) –2.601(8) Å] and close to the values [2.59(8)–2.62(8) Å] reported by Brown & Beerntsen (1965). The refined occupancy (sof) of the additional Nb2 atoms (7.1%) is almost equal to that observed in the previous 2H structure with the stacking sequence AbAδBcBαCaC β (A, B, C refer to the selenide layers; a, b, c to the completely filled niobium layers and α, β, δ to the partially filled niobium layers).
The octahedrons Nb2Se6 are slightly distorted with three short [2.288(10) Å] and three long [2.698(14) Å] distances. The Nb2 atoms are then shifted from the center of the octahedrons in order to minimize the Nb1-Nb2 [3.425(17) Å] interactions.
In order to avoid the direct stacking Nb1—Nb2—Nb1, a relaxation occurs following the transition 2H-3R model and consequently the Nb1-Nb2 distance increases to 3.425(17) Å. Indeed, in the 2H structure the relaxation by introducing high concentration of vacancy in the filled Nb layers may destabilize the structure by breaking the Nb-Nb bonding across the face sharing polyhedrons.
In the 3R-Nb1.085Se2 (Fig. 1B), the refinement reveals a high occupancy, about 11.8%, of additional Nb2 atoms. By comparing with the previous 3R structure, the Nb2Se6 octahedrons are less distorted Nb2-Se = 2.362(11)-2.621(14) Åand the Nb1-Nb2 distance (3.36(2) Å) becomes shorter (Fig. 2B). This can be explained by a shrink of the Nb-Nb layers with high content of extra Nb2 atoms.
In this case, both models exhibit relaxation: 2H-3R transition followed by a defect model introducing about 3% of vacancy in the Nb filled layers. A comparable defect was observed in the 3R-Nb1.06S2 (Powell & Jacobson, 1981).
The composition obtained with the structure refinement is: 3R-(Nb0.97(2) Δ0.03) Nb0.118(13)Se2 (Δ denotes Nb vacancy in the fully layers) (ie) 3R-Nb1.085Se2.
In Nb1+xSe2, as in the layered transition metal dichalcogenides, vacancies in the sublattice of the metal (Nb) and chalcogen (Se) are expected.
It is quite clear that the increase in the additional Nb induces changes in the pressure of saturated Se vapor above the surface of the growing crystal, and then affects the concentration of selenium vacancies. However, a free refinement of the occupancy of Se atoms in the three polytypes leads to a fully occupied sites, possibly due to the excess of Se used as starting material. Therefore, the generation of vacancies in the sublattice of Nb should be correlated to the location of Nb interlayer.
By the presence of extra Nb atoms, a significant residual electron densities are observed in the crystal structure refinements stacking in a way that violates the ideal 2H (ABAB..) and 3R (ABCABC..), about 7.3% for the former and 7.1–11.8% for the latter. The remaining 92.7% and 92.9–88.2% are unfaulted (see CIFs in Electronic Supplemental Files). Certainly, a stacking fault is a common feature in this kind of layered structures, with strong diffuse scattering observed along the c∗ axis in electron diffraction patterns (see Fig. S1).
An indication about the relative stability of 2H and 3R-Nb1+xSe2 structures, can be obtained by comparing the variation of c/a and (c/n)/a ratios with x, where a and c are the unit cell parameters and n the number of layers in the stacking sequence; n = 2 and 3 for 2H and 3R respectively.
As shown in Fig. 3, this ratio change is relatively small with the increase of x for both polytypes with a discontinuity at the 2H to 3R transition around x = 0.07. It is assumed that both polytypes may co-exist in the range 0< x < 0.07, and above this limit only the form 3R predominates. Comparable results were observed in the study of the non-stoichiometric 2H and 3R-Nb1+xS2 (0.07 < x < 0.18) by powder diffraction (Fisher & Sienko, 1980). The mechanism involving the 2H-3R transition is still unclear, and may be attributed to the phase limit of the non-stoichiometric Nb1+xSe2. The difference in energy between polytypes is small, thus the possibility to obtain a mixture of polytypes by changing the conditions of synthesis cannot be ruled out.
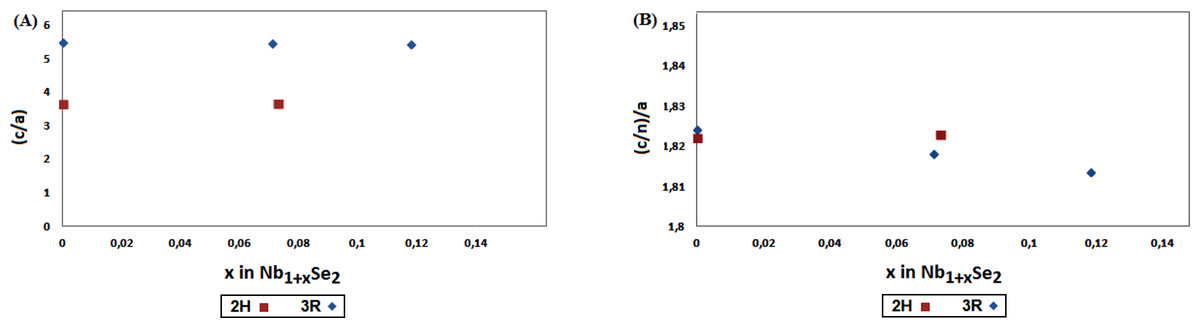
Figure 3: The reduced lattice parameters ratio (A) c/a; (B) (c/n)/a for 2H and 3R-Nb1+xSe2 (0 < x < 0.1) polytypes.
The values at x = 0 for 2H and 3R are calculated respectively from the references (Brown & Beerntsen, 1965; Meerschaut & Deudon, 2001).{kind=link}
The present results are in agreement with earlier studies by Huisman, Kadijk & Jellinek (1970) and Selte, Bjerkelund & Kjekshus (1966). The mixture 2H and 3R polytype were prepared at temperature of 760 °C in the range 0.07 < x < 0.118, yet the 3R phase at x = 0.07 required higher temperature 1,050−1100 °C. Further experimental and theoretical works are mandatory to determine the x range of 2H-3R transition accurately.
Theoretical results
To corroborate the single X-ray conclusions, the stability of four structures: the pure 2H- NbSe2 and 3R- NbSe2, 2H-Nb1.1Se2 and 3R-Nb1.1Se2 with 10% of extra Nb-SI in the pure 2H and 3R, respectively, have been investigated. The optimized structural parameters of the supercell structures are summarized in Table 2.
| Polytype | a , c (Å) | c/a | (c/n)/a | Nb1–Se , Nb2–Se (Å) | δ (Å) | ΔE | ΔG1033,1323 |
|---|---|---|---|---|---|---|---|
| 2H-NbSe2 2H-Nb1.1Se2 3R-NbSe2 3R-Nb1.1Se2 |
a = 3.463 , c = 13.210 a = 3.470 , c = 13.807 a = 3.506 , c = 19.708 a = 3.412 , c = 21.679 |
3.814 3.979 5.621 6.300 |
1.907 1.990 1.873 2.100 |
2.587–2.600 2.569-2.687, 2.564-2.579 2.597–2.600 2.592–2.697, 2.610–2.797 |
0.013 0.118 , 0.195 0.03 0.105 , 0.187 |
−1.11 - 0.93 −1.25 −0.94 |
−0.96 , −0.97 −0.97 , −0.98 |
Notes:
Note: Same notation for Nb and Se atoms as used in X-ray refinement tables. The distortion δ (Å) is the 6 difference between the longest and shortest Nb-Se bond distance.
The calculated c lattice parameters of 2H and 3R-NbSe2 are slightly overestimated compared to the experimental values by 5.28% and 4.38% respectively. This discrepancy is due to the failure of GGA (local and semi-local approximations for the exchange–correlation) in describing the van der Waals interactions.
Structural geometrical optimization revealed that additional Nb2 atoms incorporated in 2H and 3R-Nb1+xSe2 with x = 0.1, obviously increase the c parameter by 7.61% and 9.73% respectively while the a parameter remains almost constant.
The Nb1-Se distance slightly increases in pure 2H and 3R-NbSe2 (2.587–2.600 Å and 2.597–2.600 Å) compared to 2H and 3R-Nb1.1Se2 (2.569 –2.687 Å and 2.592–2.697 Å) while the Nb2-Se distances range from 2.564–2.759 Å and 2.610–2.797 Å respectively.
The calculated formation energies at T = 0 K (Table 2) indicate that the four structures polytypes can be thermodynamically stable. However, due to the strong Nb-Nb interactions the Nb (x = 0.1) SI system is found to be the least favorable. The close free energy ΔG values at high temperature suggest that facile phase transition between the two polytypes may occur, as established between Nb2Se3 and Nb1.33Se2 polytypes Ivanova et al. (2019). These results can explain the experimental stability of both 2H and 3R between 0 < x < 0.07, and confirm the hypothesis of a phase transition from 2H to 3R beyond x = 0.1.
The band structures and density of states (PDOS) of the foregoing four cases have been presented to study the impact of the Nb-SI on the electronic structure for both 2H and 3R-NbSe2.. Figures 4A and 4B show the band structure of the pure 2H and 3R respectively. The presence of several bands across the Fermi level EF reveals the metallic nature of the pure polytypes.
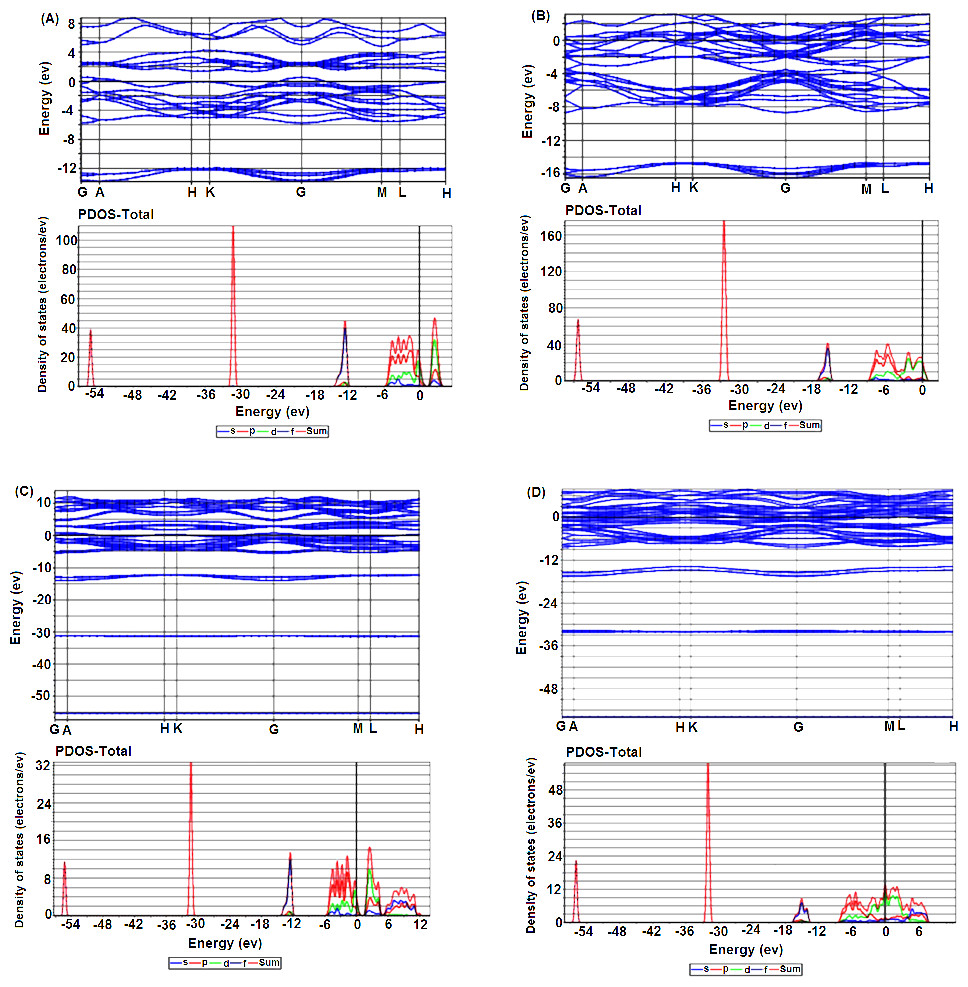
Figure 4: Electronic band structure and their corresponding DOS of (A) 2H-NbSe2; (B) 2H-Nb1.1Se2, (C) 3R-NbSe2; (D) 3R-Nb1.1Se2.
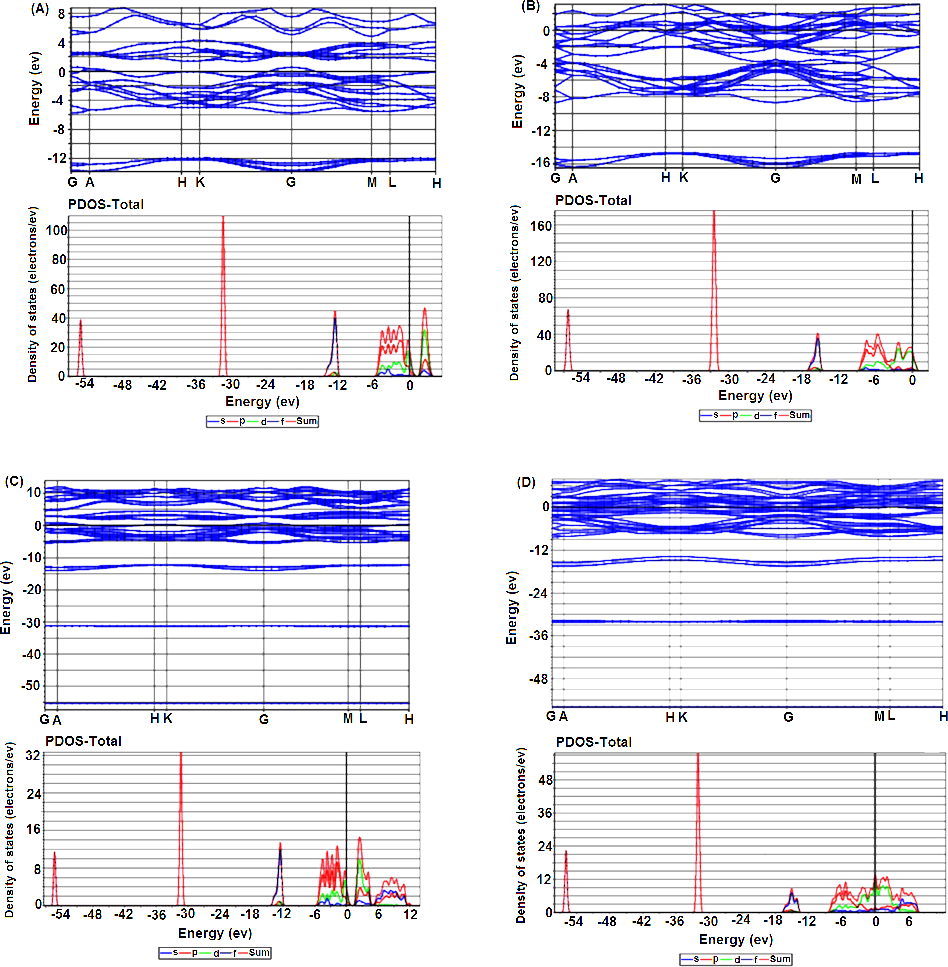{kind=link}
Partial density of states (PDOS) (see Figs. S2A and S2C), indicates that the major contribution in DOS at EF comes from the hybridization between the Nb-4d and Se-4p orbitals which is responsible for the covalent Nb-Se bonds.
For both 2H and 3R Nb-SI, the number of electronic bands around EF apparently increases compared to the pure 2H and 3R (Figs. 4C and 4D), and consequently the gap around 2 eV in the Conduction Band disappears. Moreover, the PDOS at EF decreases and upshifts (see Figs. S2B and S2D) compared to the pure polytypes which implies that a large degree of electrons is transferred by Nb extra atoms into these pure polytypes.
It is worth noticing, that the PDOS of Nb-4d is rather broad and almost located at the same energy with Se-4p, suggesting strong interactions. These phenomenon have been observed in some doped and intercalated 2H-NbSe2 (Chen et al., 2014; Hongping et al., 2014a; Hongping et al., 2014b; Kouarta, Zanat & Belkhir, 2019; Pervin et al., 2020; Xiao-Chen et al., 2020).
Conclusions
In conclusion, the non-stoichiometric polytypes 2H-Nb1.031Se2, 3R-Nb1.071Se2 and 3R-Nb1.085Se2 have been successfully synthesized and investigated by single crystal X-ray diffraction. Although the form 3R predominates for values of x greater than 0.07, both 2H and 3R polytypes may co-exist in the range 0 < x < 0.07. A transition 2H to 3R polytypes, followed by model vacancies in the host Nb sublattice should take place in order to minimize the Nblayer—Nbextra—Nblayer repulsions between these adjacent slabs.
The calculated formation energies of 2H and 3R-Nb1+xSe2 (x = 0, 0.1) by DFT, indicate that both pure and Nb-SI systems can be thermodynamically stable, and suggest an easy phase transition between polytypes. The theoretical outcomes reveal the metallic nature of these polytypes with an overwhelming number of electrons transferred by Nb extra atoms into pure polytypes.
Further work involving experimental and theoretical investigations on the Nb-Se system is needed to elucidate the 2H-3R transition mechanism.
Supplemental Information
Atomic coordinates, Anisotropic displacement parameters, Selected bond distances for the 03 polytypes. Zone axis SAED pattern of 3R-Nb1.085Se Partial DOS
CIF of 2H-Nb1.031Se2
Raw and processed experimental data, crystal structure solution and refinement, chemical structure and connectivity for the 2H-Nb1.031Se2 polytype.
CIF of 3R-Nb1.071Se2
Raw and processed experimental data, crystal structure solution and refinement, chemical structure and connectivity for the 3R-Nb1.071Se2 polytype.
CIF of 3R-Nb1.085Se2
Raw and processed experimental data, crystal structure solution and refinement, chemical structure and connectivity for the 3R-Nb1.085Se2 polytype.
DFT of 2H-Nb1Se2
Structure, Band structure, DOS, PDOS for the 2H-NbSe2 polytype, and *.CASTEP where the *.CASTEP file contains the numerical output of a CASTEP job in ASCII format. The header describes the settings used, followed by the calculation results.
DFT of 2H-Nb1.1Se2
Structure, Band structure, DOS, PDOS for the 2H-Nb1.1Se2 polytype, and *.CASTEP where the *.CASTEP file contains the numerical output of a CASTEP job in ASCII format. The header describes the settings used, followed by the calculation results.
Structure, Band structure, DOS, PDOS for the 3R-NbSe2 polytype, and *.CASTEP where the *.CASTEP file contains the numerical output of a CASTEP job in ASCII format
The header describes the settings used, followed by the calculation results.
Structure, Band structure, DOS, PDOS for the 3R-Nb1.1Se2 polytype, and *.CASTEP where the *.CASTEP file contains the numerical output of a CASTEP job in ASCII format
The header describes the settings used, followed by the calculation results.