
Polysulfide-assisted urea synthesis from carbon monoxide and ammonia in water
- Published
- Accepted
- Received
- Academic Editor
- Eder Lenardao
- Subject Areas
- Green Chemistry, Organic Chemistry (other), Synthetic Organic Chemistry
- Keywords
- Carbonyl sulfide, Hydrogen sulfide, Green chemistry, Sustainable chemistry
- Copyright
- © 2022 Kitadai et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Organic Chemistry) and either DOI or URL of the article must be cited.
- Cite this article
- 2022. Polysulfide-assisted urea synthesis from carbon monoxide and ammonia in water. PeerJ Organic Chemistry 4:e6 https://doi.org/10.7717/peerj-ochem.6
Abstract
Efficient conversion of carbon monoxide into urea in an aqueous ammonia solution was demonstrated through coupling with the elemental sulfur reduction to polysulfides. This reaction starts with a simple mixture of carbon monoxide, ammonia, elemental sulfur, and a small amount of hydrogen sulfide for polysulfide formation, enabling an almost complete conversion of 1 atm of carbon monoxide to urea (95–100% yield) within 216, 64, and 32 h at 35 °C, 50 °C, and 65 °C, respectively. Polysulfides control the overall reaction rate while suppressing the accumulation of a by-product, hydrogen sulfide, to less than 1 Pa. These functions follow simple kinetic and thermodynamic theories, enabling prediction-based reaction control. This operational merit, together with the superiority of water as a green solvent, suggests that our demonstrated urea synthesis is a promising option for sulfur utilization beneficial for agricultural production.
Introduction
Sulfur is a crucial element in agriculture. Besides being a vital nutrient for crop production, sulfur is a common pesticide in the form of elemental sulfur (S0) (Griffith, Woodrow & Seiber, 2015). S0 is also used as a coating material of urea to regulate urea availability to plants (Naz & Sulaiman, 2016; Wesolowska et al., 2021), while its oxidation to sulfate (SO42−) is applicable to control soil pH (Roig, Cayuela & Sanchez-Monedero, 2004). Moreover, sulfuric acid is the mostly used leaching reagent for phosphate extraction from phosphorites (Sahu et al., 2014; Wagenfeld et al., 2019).
Worldwide, the majority of anthropogenic sulfur is derived from coal and petroleum refinements (Wagenfeld et al., 2019; Rappold & Lackner, 2010). Although the global sulfur supply currently exceeds the market demand (Rappold & Lackner, 2010; Srivastava, 2012; Saleh, 2020), replacement of fossil fuels with renewable energy sources is expected to lead to a sulfur shortage in the future (Wagenfeld et al., 2019). For sustainable agricultural activity involving sulfur, whose market price possibly increases due to its dwindling supply, an economically viable option of sulfur valorization beneficial for agriculture ought to be explored. Such sulfur valorization should also be important in the current sulfur oversupply situation.
In this article, we demonstrate polysulfide-assisted urea synthesis from carbon monoxide (CO) and ammonia (NH3) in water. Urea accounts for approximately 50 wt% of the global nitrogen consumption as fertilizer (Glibert et al., 2006; IFASTAT, https://www.ifastat.org/). Its production worldwide was over 180 Mt in 2020, and is expected to grow over the next few decades in accordance with the increasing global food demand (Erisman et al., 2008; Bodirsky et al., 2014). Conventionally, industrial urea synthesis is based on the reaction of carbon dioxide (CO2) with NH3 at high temperatures (170–200 °C) and pressures (130–250 bar). Because multi-stage cyclic systems are required to improve the otherwise low reaction efficiency, the overall process is energy consuming, emitting higher amounts of CO2 than those converted to urea (Bargiacchi, Antonelli & Desideri, 2019; Zendehboudi et al., 2014; Rafiee et al., 2018). CO is a promising alternative to CO2 because of its superior reactivity (Diaz, Darko & McElwee-White, 2007) and growing industrial availability. Highly efficient CO production is now realized on various low-cost electrocatalysts (Satanowski & Bar-Even, 2020). The high amount of CO produced through methane steam reforming (Giddey, Badwal & Kulkarni, 2013; Smith, Hill & Torrente-Murciano, 2020) may also be a practical choice.
There are several reports on the synthesis of urea and urea derivatives from CO and NH3 or amines with the aid of S0 (Franz & Applegath, 1961; Franz et al., 1961a, 1961b; Mizuno et al., 2006, 2007; Mizuno, Nakai & Mihara, 2009; Peng, Li & Xia, 2006). However, experiments have typically been conducted in organic solvents; to the best of our knowledge, there are no experimental studies on urea synthesis in water. A difficult intermediate step in water is the formation of thiocarbamate. In certain organic solvents (e.g., N,N-dimethylformamide), thiocarbamates have been shown to form through the binding of the corresponding amines to S0 via the S–S bond cleavage, followed by nucleophilic attack of CO to the resultant thiolate species (Mizuno et al., 2006, 2007; Mizuno, Nakai & Mihara, 2009). No thiocarbamate has been formed in this manner when water was used as a solvent (Mizuno, Iwai & Ishino, 2005).
In alkaline aqueous solutions, polysulfides (PSs) provide an alternative route to thiocarbamate (Scheme 1). PSs are dissolved sulfur chains (Sn2−; n = 2–8) formed from the reaction of S0 with bisulfide (HS−) (Avetisyan, Buchshtav & Kamyshny, 2019). Because sulfur atoms in PSs are electrophilic at the non-terminal position (Fukuto et al., 2018), PSs are susceptible to nucleophilic attack by CO to form carbonyl sulfide (OCS) (Kamyshny et al., 2003). OCS is known to readily react with amines to form thiocarbamates, and facilitate a variety of aqueous organic processes (Huber & Wachtershauser, 1998; Leman, Orgel & Ghadiri, 2004, 2006). We will show below that these steps occur consecutively in water from a simple mixture of CO, NH3, S0, and a small amount of HS−. Under moderately alkaline pH (10.3–10.5) and temperature (35 °C, 50 °C or 65 °C), an almost complete conversion of 1 atm of CO to urea (95–100% yield) was achieved in the presence of excess amounts of NH3 and S0. Importantly, PSs control the rate of OCS formation (Kamyshny et al., 2003), which determines the overall rate of urea production. Moreover, PSs suppress the accumulation of HS– by-product through the PS–HS− equilibrium. Thus, the timescale of urea formation and the resultant H2S concentration can be predicted from kinetic and thermodynamic calculations by parameterizing the PS concentration.
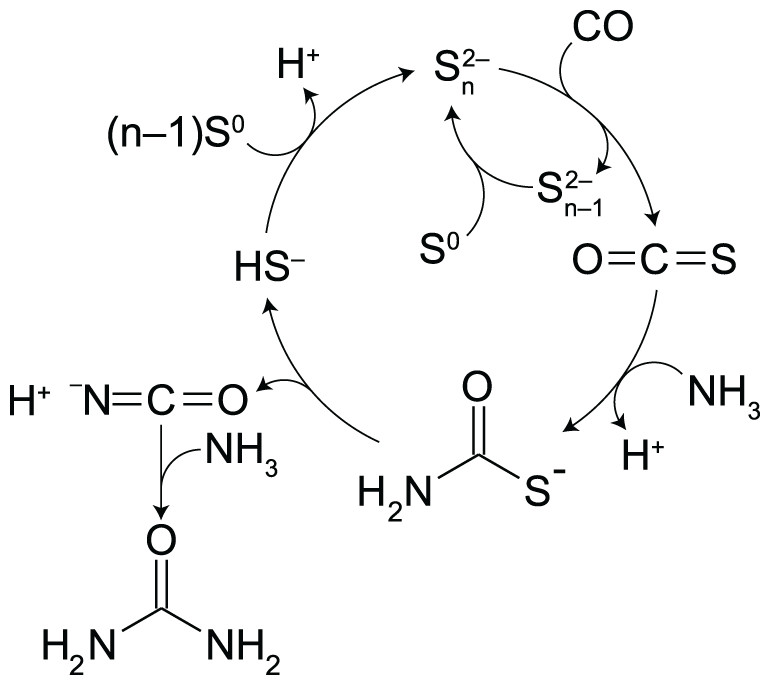
Scheme 1: Proposed intermediate steps in the polysulfide-assisted urea synthesis from CO and NH3 in water.
{kind=link}
Note that PSs are widely used corrosion inhibitors in the petrochemical industry (Tiu & Advincula, 2015). In the presence of oxygen (O2), PSs are oxidized to S0 at approximately four times the rate of HS– oxidation (Steudel, Holdt & Nagorka, 1986; Kleinjan, de Keizer & Janssen, 2005). Although the PS–O2 reaction also generates thiosulfate (S2O32−) as a by-product, its further oxidation to sulfate (SO42−) readily proceeds with the aid of sulfide catalysts (e.g., CuS; Lara et al., 2019) or by the action of ultraviolet light (Ahmad et al., 2015). SO42− is a sulfur species directly available to plants (Lucheta & Lambais, 2012; Griffith, Woodrow & Seiber, 2015; Fuentes-Lara et al., 2019). Thus, sulfur is not a mere oxidant facilitating selective urea synthesis from CO and NH3 under mild and non-corrosive conditions. The resultant aqueous suspension is potentially applicable as a sulfur and nitrogen fertilizer source.
Materials and Methods
Urea synthesis experiments were conducted in a serum bottle filled with CO (0.33 mmol, 1 atm), S0 (3.1 mmol), and 5 mL of an aqueous solution of NH3 (14.5 mmol), ammonium chloride (NH4Cl, 1.45 mmol), and sodium hydrogen sulfide (NaHS, 32.5 μmol). NH4Cl was used at a molar ratio of NH4Cl/NH3 = 0.1 to keep the pH at 10.3–10.5 (in the absence of NH4Cl, the pH was 10.7–10.9 after the urea synthesis experiment). NaHS was added for the formation of PSs ((n − 1)S0 + NaHS → Sn2− + Na+ + H+) at a concentration of 6.5 mM, which corresponded to 10% of the initial amount of CO. All the reactions involving CO were carefully carried out in a fume hood or a glove box with strict precautions. For more details and product analysis methods, see Materials & Methods in Supplemental Information.
Results and discussion
Experimental results
Figure 1A shows the yield of urea, based on CO, at three different temperatures (i.e., 35 °C, 50 °C, and 65 °C). Urea formation was faster at higher temperatures, reaching 80% yield within 24 h at 65 °C, 48 h at 50 °C, and 168 h at 35 °C. Longer reaction duration led to further urea formation; 95–100% yield of urea was obtained after the 32-, 64-, and 216-h reaction at 65 °C, 50 °C, and 35 °C, respectively (Fig. 1A). Similar time and temperature dependencies were observed at an NH3 concentration of 5.6 M (Fig. S5). CO2 was formed as a by-product, but the yield was at most 0.8% under the examined reaction conditions. When no NH4Cl was added as a starting material, the urea selectivity slightly decreased, while CO2 formed in higher amount, compared with those in the presence of NH4Cl under otherwise identical condition. For example, the 120-h reaction at 50 °C in the absence of NH4Cl resulted in 94 ± 5% yield of urea and 2 ± 0.2% yield of CO2. No urea was detected when either NH3 or S0 was absent and when CO was replaced with CO2. In the absence of NaHS, the reaction started with a sluggish urea-formation stage (green dotted line in Fig. 1A). This induction period was followed by an accelerated growth of the urea yield that eventually exceeded 90%. The mechanism underlying the sigmoidal-like growth is discussed later.
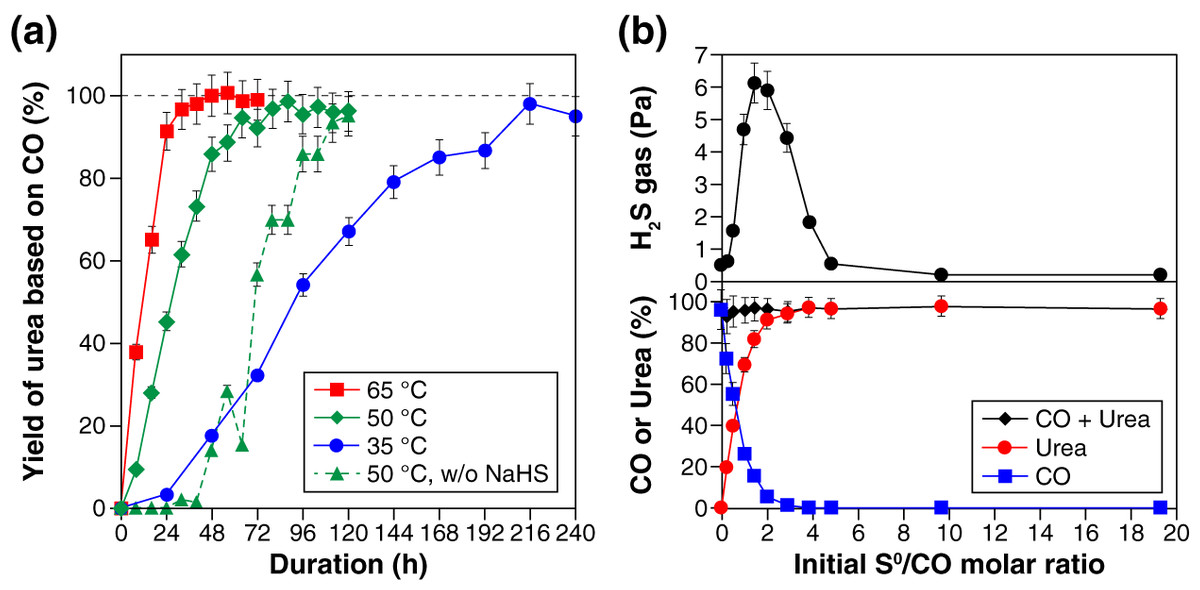
Figure 1: Experimental results for the sulfur-assisted urea synthesis.
(A) The yield of urea at 35 °C, 50 °C, and 65 °C. The results obtained without NaHS as a starting material are also shown with triangle symbols connected by dotted lines. (B) Effect of the initial S0/CO molar ratio on the H2S gas concentration (top), the yield of urea (bottom), and the amount of CO remaining (bottom) after the 120-h reaction at 50 °C. In this experiment, the initial amount of S0 was varied between 0 and 6.2 mmol, while that of CO was kept constant at 0.33 mmol.{kind=link}
The initial amount of S0 had a strong impact on the concentration of H2S by-product, as well as the yield of urea, as seen in the results obtained after the 120-h reaction at 50 °C (Fig. 1B). With an increase in the S0/CO molar ratio from 0 to 19.1, the H2S gas concentration initially exhibited a rapid increase to approximately 6 Pa, followed by a decrease to a value less than 1 Pa at the S0/CO molar ratio of 4.8, and then reached a steady value (ca. 0.2 Pa) at higher S0/CO molar ratios. As for urea, the yield increased significantly when the S0/CO molar ratio was between 0 and 2 at the expense of CO (Fig. 1B).
The amount of S0 always decreased throughout the reaction; few or no S0 remained in the experiments with the initial S0/CO molar ratio less than 4.8 (Fig. S6). Meanwhile, the aqueous solutions exhibited a yellow color characteristic of PSs (Bedoya-Lora, Hankin & Kelsall, 2019); darker yellow was observed when larger amount of S0 was used (Fig. S6). These features indicate that S0 was reduced to PSs coupling with CO oxidation to urea (Eq. (1)) and CO2 (Eq. (2))
(1)
(2)
Indeed, the molar amount of PSs formed through the reaction exhibited a one-to-one correlation with the amount of CO consumed (Fig. S7).
Thermodynamic calculation
Thermodynamic calculation predicts that in the presence of S0, PSs are the dominant dissolved S species over HS− and H2S at the examined pH (10.3–10.5) (Fig. 2A). The presence of PSs with S0 thus suppress the accumulation of H2S by-product in the gas phase, keeping the H2S gas concentration below 0.5 Pa in our experimental setting (Fig. 2B). When no PS formation was assumed, two orders of magnitude higher values (13–21 Pa) were calculated for the steady-state H2S gas concentration after the complete conversion of CO to urea or CO2 (Fig. 2B). Because the mean chain length of PSs is 5.0 (i.e., n = 5 in Sn2−, Fig. 2A), the capability of PSs to suppress H2S accumulation is expected to work effectively at S0/CO molar ratios higher than five. When an insufficient amount of S0 is used (e.g., S0/CO molar ratio = 2), in contrast, the S0-to-PSs reduction cannot fully extract the reduced sulfur (S0 → S2−), resulting in an elevated emission of H2S gas (Fig. 1B). Thermodynamic calculations taking PSs into account thus explain our experimental results for H2S. At the reaction temperatures of 35 °C, 50 °C, and 65 °C, the gas-phase H2S are expected to reach 0.3–0.8, 1.4–3.0, and 4.7–9.8 Pa, respectively.
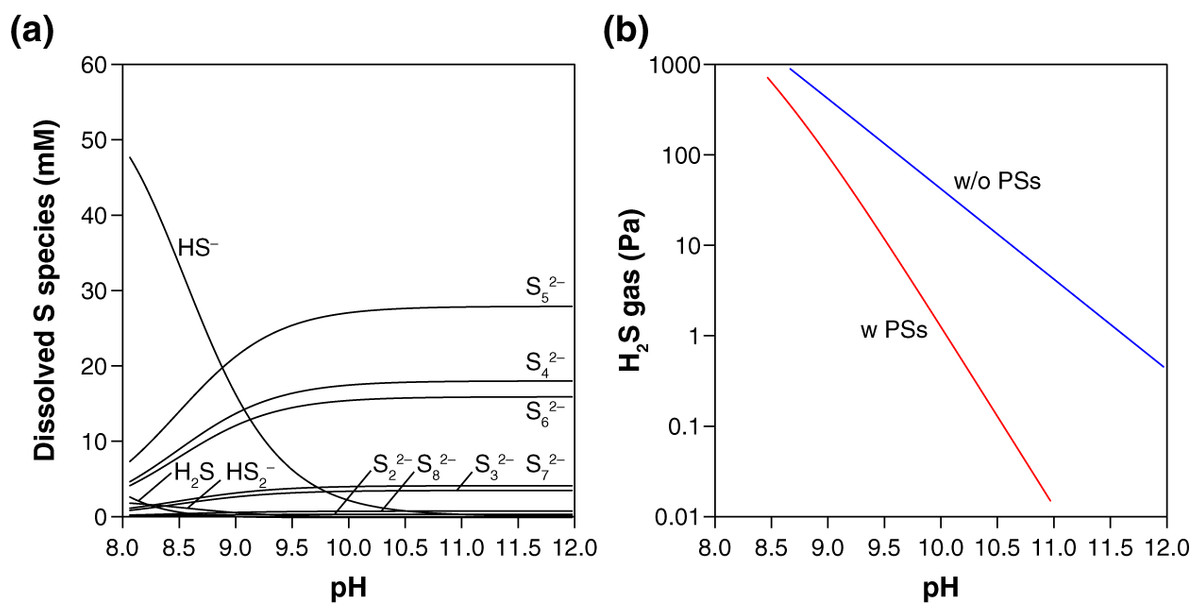
Figure 2: Thermodynamic prediction for the equilibrium concentrations of dissolved sulfur species (A) and gas-phase H2S (B) after the urea synthesis reaction.
In these calculations, the presence of an excess amount of S0 for a complete conversion of CO (0.33 mmol) to urea or CO2 was assumed (i.e., the initial S0/CO molar ratio ≫ 5; see the section “Thermodynamic calculation”). The blue line in (B) was calculated excluding PSs from the system. All the calculations were preformed using the experimentally derived thermodynamic data for PSs, HS−, H2S reported in the literature (Shock et al., 1997; Shock, Helgeson & Sverjensky, 1989; Robie & Hemingway, 1995; Kamyshny et al., 2004, 2007).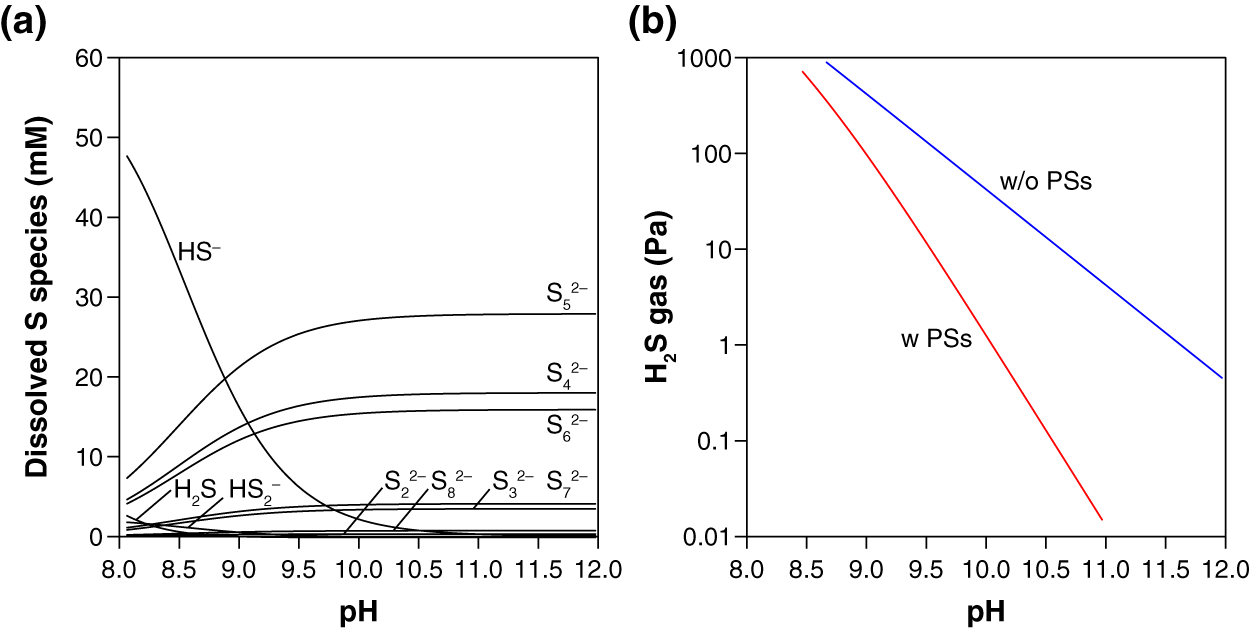{kind=link}
Based on the thermodynamic calculation (Fig. 2A), the urea synthesis reaction can be simplified as:
(3)
On average, five atoms of S0 were converted to a single molecule of S52− when one molecule of CO was consumed to form urea.
Kinetic characteristics
In water, PSs readily react with CO to form OCS (Kamyshny et al., 2003), a likely key intermediate in PS-assisted urea synthesis (Scheme 1). OCS was indeed observed in our experiments as a trace and transient gas species (Fig. S8). The formation rate of OCS is linearly proportional to the concentrations of CO and PSs (Kamyshny et al., 2003). Once formed, the OCS-to-urea conversion proceeds in competition with the OCS hydrolysis to CO2, whose half-life is in the seconds-to-minutes range under the examined reaction conditions (e.g., 18 s at pH10.0 and 50 °C; Kamyshny et al., 2003). Because CO2 was a minor product in our experiments although the rate of OCS hydrolysis is much higher than that observed for urea synthesis (Fig. 1A), OCS formation is expected to be the rate-determining step of the overall urea synthesis process.
In accordance with this kinetic consideration, the experimental results (Fig. 1A), together with the corresponding CO consumption, were well represented with the following second-order rate equation parameterizing the CO and PS concentrations (Figs. 3A–3C ):
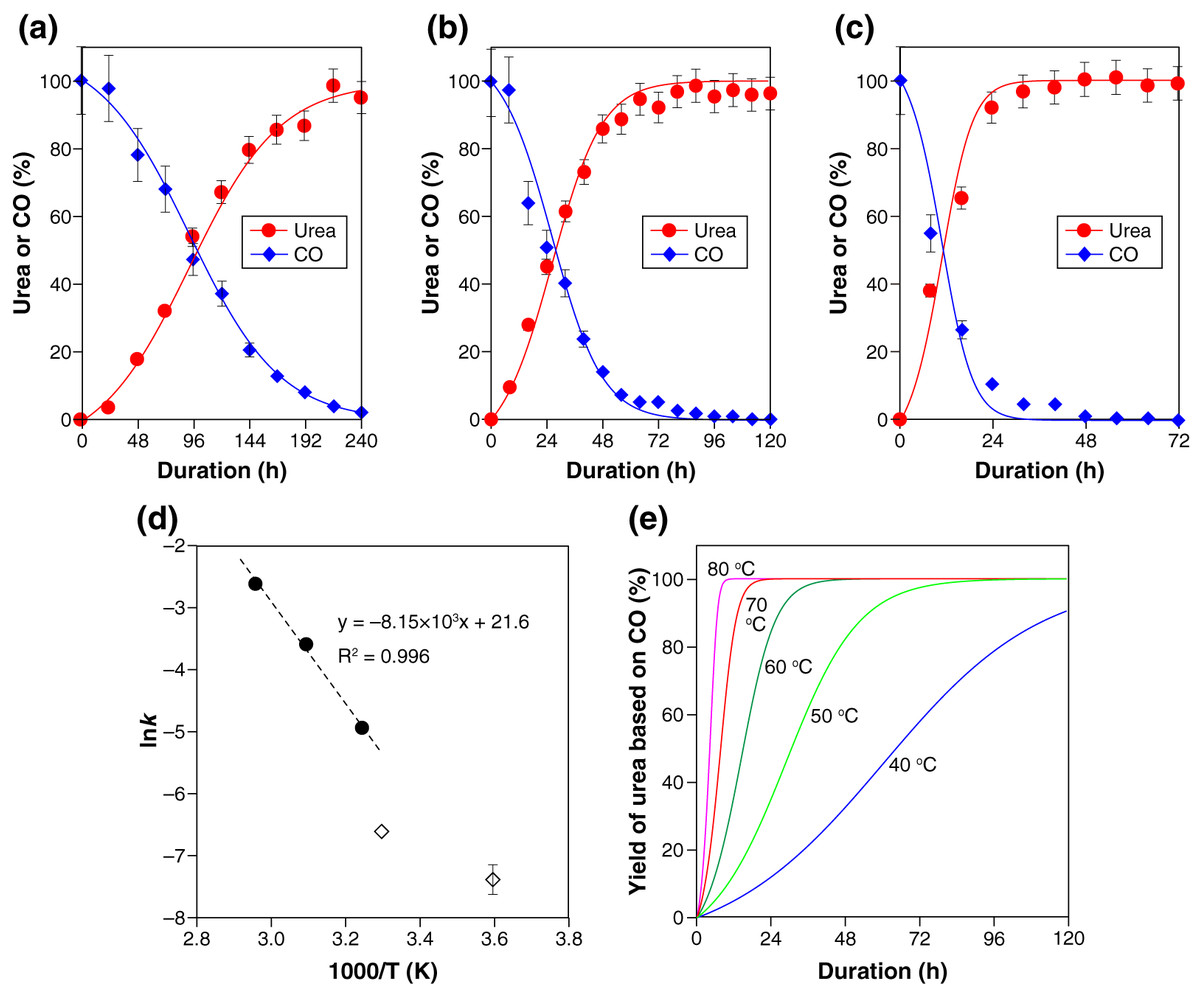
Figure 3: Kinetic characteristics of the polysulfide-assisted urea synthesis.
(A–C) The experimental data (closed symbols) and fitted results (solid lines) for the CO-to-urea conversion at 35 °C (A), 50 °C (B), and 60 °C (C). The lines were calculated with Eqs. (4)–(6) by setting the rate constant k to 0.0072 (A), 0.028 (B), or 0.075 (C) mol−1 L−1 s−1. The experimental data for urea are also shown in Fig. 1A. (D) Arrhenius plot of the obtained rate constants (closed circles). The reported rate constants for OCS formation from CO and PSs (Kamyshny et al., 2003) are also shown (open diamond symbols). (E) The yield of urea as a function of time between 40 °C and 80 °C calculated with the activation energy (Ea = 67.8 kJ mol–1) and pre-exponential factor (ln(A) = 21.6) obtained from the Arrhenius plot (D).{kind=link}
(4) where [x] denotes the concentration of species x in water. Note that the urea-formation reaction (Eq. (1)) generates an equimolar amount of PSs while consuming an equimolar amount of CO. PSs and CO thus has the following kinetic relationships with urea in this reaction:
(5)
(6)
In Eq. (6), the time derivative of the total amount of CO ([CO]Tot) is correlated with that of urea concentration multiplied by the volume of sample solution (VL), because CO is distributed in both the gas and aqueous phases. Thus, as long as competing reactions (e.g., Eq. (2)) are not significant, CO and PSs do not need to be monitored during the experiment for the use of Eq. (4). Considering the high OCS hydrolysis rate at high pH (Kamyshny et al., 2003), a moderately alkaline pH (e.g., pH 10–11) is preferable for such selective urea synthesis. It is also suggested from the adequate kinetic representation (Figs. 3A–3C) that the other intermediate species (i.e., thiocarbamate and isocyanate; Scheme 1) occupy a tiny fraction of the carbon compounds present in the course of the reaction.
Equations (4)–(6) also enabled us to reproduce the experimental results obtained without NaHS as the starting material (Fig. S9). However, this simulation requires the initial PS concentration to be a non-zero value (0.18 mM; see the caption of Fig. S9 for the other parameters). This low PS concentration is likely derived from the hydrolysis of S0 (4S0 + 4H2O → 3HS– + SO42− + 5H+) (Ellis & Giggenbach, 1971) followed by the formation of PSs from S0 and HS− ((n−1)S0 + HS− → Sn2− + H+).
The determined rate constants k at three reaction temperatures (35 °C, 50 °C, and 65 °C) exhibited a linear trend in the Arrhenius plot (Fig. 3D). Extrapolation of the regression line to lower temperatures led to values roughly consistent with the reported rate constants for OCS formation from CO and PSs (diamond symbols in Fig. 3D) (Kamyshny et al., 2003).
The activation energy (Ea = 67.8 kJ mol−1) and pre-exponential factor (ln(A) = 21.6) obtained from the Arrhenius plot (Fig. 3D) enables us to predict the urea yield as a function of time in a range of temperature. For example, 80% yield of urea is calculated to be achieved within 98, 47, 23, 12, and 6 h at 40 °C, 50 °C, 60 °C, 70 °C, and 80 °C, respectively (Fig. 3E). Such estimations, together with the thermodynamic calculation for H2S exemplified above, should be an important basis for realizing urea manufacturing with maximum productivity while minimizing the emission of H2S gas.
Versatility of the PS-assisted reaction mechanism
Several techniques are available for the isolation and purification of urea from aqueous solution, such as forward osmosis (Ray, Perreault & Boyer, 2019), the use of adsorbents (e.g., activated carbons, zeolites, ion-exchange resins, and silica) (Urbanczyk, Sowa & Simka, 2016), and selective dissolution in certain solvents (e.g., ethanol) and subsequent recrystallization (Marepula, Courtney & Randall, 2021). Methods have also been developed for NH3 recovery from wastewaters (Kuntke et al., 2018; Kim, Moreno-Jimenez & Efstathiadis, 2021). PSs is oxidized by O2 to S0 (Steudel, Holdt & Nagorka, 1986; Kleinjan, de Keizer & Janssen, 2005), which may be used for the next round of urea production. Urea can be isolated from dissolved inorganic species by utilizing its higher solubility in organic solvents (Marepula, Courtney & Randall, 2021). When ethanol is considered as a solvent for urea isolation from our product solution, the optimum purity of 88.2 mol% is expected based on the reported solubility data of urea and ammonium, sodium, and sulfate salts (Lee & Lahti, 1972; Toro, Dobrosz-Gomez & Garcia, 2014; Galvao et al., 2021). Experimental test of the isolation process and its further development must be a crucial next step for urea commercialization. It is also noteworthy that in addition to urea, NH3, S0, and SO42− (a by-product of PS oxidation) are well-used fertilizer components. Thus, not only as an isolated form, urea as a mixture component with the other N and S species could also be useful in agriculture (Wagenfeld et al., 2019).
The versatility of sulfur in water demonstrated in this study should also be beneficial to the production of various urea derivatives serving as intermediates for fine chemicals, pharmaceuticals, cosmetics and pesticides (Franz & Applegath, 1961; Franz et al., 1961a, 1961b; Mizuno et al., 2006, 2007; Mizuno, Nakai & Mihara, 2009; Peng, Li & Xia, 2006); thus, possessing wide applicability in material manufacturing. The reaction mechanism may also have played a role in prebiotic chemical processes in primordial hydrothermal vent environments rich in sulfur, CO and NH3 that eventually led to the origin of life (Li et al., 2017; Li, Kitadai & Nakamura, 2018; Nakashima et al., 2018; Lee et al., 2021; Kitadai, Kameya & Fujishima, 2017; Kitadai et al., 2018, 2019, 2021).
Conclusions
In summary, we have reported selective CO conversion to urea in a mild aqueous ammonia solution assisted by PSs, and elucidated the thermodynamic and kinetic characteristics of this reaction. This simple and environmentally benign aqueous process should be worth considering as a green and sustainable application of sulfur that is beneficial to agricultural activity.